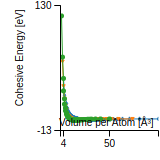
Citations
This panel presents information regarding the papers that have cited the interatomic potential (IP) whose page you are on.
The OpenKIM machine learning based Deep Citation framework is used to determine whether the citing article actually used the IP in computations (denoted by "USED") or only provides it as a background citation (denoted by "NOT USED"). For more details on Deep Citation and how to work with this panel, click the documentation link at the top of the panel.
The word cloud to the right is generated from the abstracts of IP principle source(s) (given below in "How to Cite") and the citing articles that were determined to have used the IP in order to provide users with a quick sense of the types of physical phenomena to which this IP is applied.
The bar chart shows the number of articles that cited the IP per year. Each bar is divided into green (articles that USED the IP) and blue (articles that did NOT USE the IP).
Users are encouraged to correct Deep Citation errors in determination by clicking the speech icon next to a citing article and providing updated information. This will be integrated into the next Deep Citation learning cycle, which occurs on a regular basis.
OpenKIM acknowledges the support of the Allen Institute for AI through the Semantic Scholar project for providing citation information and full text of articles when available, which are used to train the Deep Citation ML algorithm.
|
This panel provides information on past usage of this interatomic potential (IP) powered by the OpenKIM Deep Citation framework. The word cloud indicates typical applications of the potential. The bar chart shows citations per year of this IP (bars are divided into articles that used the IP (green) and those that did not (blue)). The complete list of articles that cited this IP is provided below along with the Deep Citation determination on usage. See the Deep Citation documentation for more information.
118 Citations (28 used)
Help us to determine which of the papers that cite this potential actually used it to perform calculations. If you know, click the .
USED (definite) S. Blackwell, R. Smith, S. Kenny, J. Walls, and C. Sanz-Navarro, “Modelling the growth of ZnO thin films by PVD methods and the effects of post-annealing,” Journal of Physics: Condensed Matter. 2013. link Times cited: 29 Abstract: Results are presented for modelling of the evaporation and m… read moreAbstract: Results are presented for modelling of the evaporation and magnetron sputter deposition of ZnxOy onto an O-terminated ZnO ( 0 0 0 1 ̄ ?> ) wurtzite surface. Growth was simulated through a combination of molecular dynamics (MD) and an on-the-fly kinetic Monte Carlo (otf-KMC) method, which finds diffusion pathways and barriers without prior knowledge of transitions. We examine the effects of varying experimental parameters, such as substrate bias, distribution of the deposition species and annealing temperature. It was found when comparing evaporation and sputtering growth that the latter process results in a denser and more crystalline structure, due to the higher deposition energy of the arriving species. The evaporation growth also exhibits more stacking faults than the sputtered growth. Post-annealing at 770 K did not allow complete recrystallization, resulting in films which still had stacking faults where monolayers formed in the zinc blende phase, whereas annealing at 920 K enabled the complete recrystallization of some films to the wurtzite structure. At the latter temperature atoms could also sometimes be locked in the zinc blende phase after annealing. When full recrystallization did not take place, both wurtzite and zinc blende phases were seen in the same layer, resulting in a phase boundary. Investigation of the various distributions of deposition species showed that, during evaporation, the best quality film resulted from a stoichiometric distribution where only ZnO clusters were deposited. During sputtering, however, the best quality film resulted from a slightly O rich distribution. Two stoichiometric distributions, one involving mainly ZnO clusters and the other involving mainly single species, showed that the distribution of deposition species makes a huge impact on the grown film. The deposition of predominantly single species causes many more O atoms at the surface to be sputtered or reflected, resulting in an O deficiency of up to 18% in the deposited film and therefore resulting in more stacking faults and phase boundaries. The methods used allow analysis of key mechanisms that occur during the growth process and give hints as to the optimum conditions under which complete crystalline layers can form. read less USED (high confidence) G. Yang, B. Liang, Q. Zhu, Y. Hu, and X. Ye, “Comprehensive Study of the Effects of Nanopore Structures on Enzyme Activity for the Enzyme Based Electrochemical Biosensors Based on Molecular Simulation.,” The journal of physical chemistry. A. 2016. link Times cited: 2 Abstract: Assembly of biocompatible nanostructures to retain the enzym… read moreAbstract: Assembly of biocompatible nanostructures to retain the enzyme activity and improve the biocatalytic ability is a decisive factor for enhancing the performance of enzyme biosensors. However, there is still a lack of molecular level understandings of the physicochemical interaction mechanism at the interface of biosensor electrodes and enzymes. Here, for the first time at molecular level, the effects of two classic biosensor electrode materials with different electrical properties and morphologies and glucose oxidase (GOD) on retaining the enzyme conformation were analyzed by molecular dynamics simulation. First, for the immobilization of GOD, the interfaces of zinc oxide (ZnO) with different electrical properties and 10 nm diameter ZnO nanopore were studied. Then, to simulate the sensing process when electric voltages are applied, positively charged gold planes and 10 nm diameter gold nanopore were investigated as well. The results showed that the nanopore structure was confirmed to be well adapted for the enzyme conformation retaining compared to the plane structure for both ZnO and gold materials, and they almost fit well with the sensitivity measurement results from many previously reported experimental studies. This study also indicates that molecular modeling of the interactions between biomolecules and functional nanostructures is helpful for developing high performance enzyme nanobiosensors. read less USED (high confidence) G. J. Soldano, F. M. Zanotto, and M. Mariscal, “Mechanochemical stability of sub-nm ZnO chains.,” Physical chemistry chemical physics : PCCP. 2016. link Times cited: 2 Abstract: Formation of monoatomic chains by axial stretching of zinc o… read moreAbstract: Formation of monoatomic chains by axial stretching of zinc oxide nanowires is investigated using molecular dynamics and supported by density functional calculations. Special focus is made on the mechanical properties of these structures. Using a state-of-the-art force field it was found that O2 species are commonly formed within the chain. This species drastically weakens the chain strength. Previous simulations, based on a pair potential, failed to predict O2 formation. Moreover, the superductility of zinc oxide nanowires observed in earlier studies, was found to be an artifact of the pair potential. Simulations revealed that the chain length before rupture (usually of 6 atoms) is independent of the nanowire diameter. The electronic structure and the charge distribution of the chains were also studied. read less USED (high confidence) D. A. Newsome, D. Sengupta, and A. Duin, “High-Temperature Oxidation of SiC-Based Composite: Rate Constant Calculation from ReaxFF MD Simulations, Part II,” Journal of Physical Chemistry C. 2013. link Times cited: 25 Abstract: Space vehicles often encounter very high temperature and har… read moreAbstract: Space vehicles often encounter very high temperature and harsh oxidative environments. To ensure proper thermal protection, layers composed of SiC and EPDM polymer are placed on the outer surface of the space vehicle. The O2 and H2O molecules are able to oxidize the SiC network, creating SiO2-type structures that may form a protective layer, while also pyrolyzing and burning the EPDM polymer, causing ablation. Reactive molecular dynamics simulations nicely complement experiment, as they provide direct observation and information to calculate physical parameters such as transport diffusivities and reaction constants. In this study, rate models were developed and molecular dynamics simulated trajectories were used to extract Arrhenius parameters that describe the initial stages of transport and kinetics of SiC oxidation by O2 and H2O and the combustion and pyrolysis of EPDM. The simulations showed that O2 was able to oxidize SiC more efficiently than H2O, with the transport activation barrier of O2 in the r... read less USED (high confidence) B. Narayanan, I. Reimanis, H.-C. Huang, and C. Ciobanu, “Radiation effects and tolerance mechanism in β-eucryptite,” Journal of Applied Physics. 2013. link Times cited: 5 Abstract: Previous studies on Li-silicates have shown that these mater… read moreAbstract: Previous studies on Li-silicates have shown that these materials are resistant to radiation damage even in extreme physical and chemical environments, and are thus promising solid-state breeder materials in fusion reactors. Here, we focus on β-eucryptite as a member of Li-Al silicate class of ceramics with potential for nuclear applications, and study the atomic-scale processes induced by radiation. Using molecular dynamics simulations based on a reactive force field, we have found that upon radiation dosage of 0.21 displacements-per-atom or less, the structure largely retains its long-range order while exhibiting (a) disordering of the Li atoms, (b) distortion of the Si and Al tetrahedra defined as the change in their oxygen-coordination number, and (c) tilting of the Si and Al tetrahedra with respect to one another. We find that Si tetrahedra that distort to SiO3 during exposure to radiation recover significantly upon thermal relaxation, and provide the mechanism for this recovery. This mechanism consis... read less USED (high confidence) S. große Holthaus, S. Köppen, T. Frauenheim, and L. C. Ciacchi, “Atomistic Simulations of the ZnO(12̅10)/Water Interface: A Comparison between First-Principles, Tight-Binding, and Empirical Methods.,” Journal of chemical theory and computation. 2012. link Times cited: 20 Abstract: We investigate the adsorption behavior of water over the zin… read moreAbstract: We investigate the adsorption behavior of water over the zinc oxide (12̅10) surface starting from single molecules up to bulk liquid by means of atomistic molecular dynamics simulations. We compare results obtained with density-functional theory, density-functional tight binding, and a recently developed reactive force field. The methods perform comparably up to the level of a single monolayer of adsorbed water, predicting only small differences in adsorption energies and, as a consequence, adsorption geometries. These lie within the error bars of typical quantum mechanical calculations performed with different exchange-correlation functionals. However, the discrepancies among the methods have a dramatic effect on the dissociation equilibria and the structuring of liquid water layers in contact with the surface. Especially the different treatment of electrostatic interactions via self-consistent atomic point charges appears to heavily influence the simulation outcomes. Critical comparisons with experimental studies and possibly ad hoc reparametrizations of the semiempirical functionals may thus be necessary to study phenomena such as dissolution or biomolecular adsorption at ZnO surfaces within statistically relevant time and size scales. read less USED (high confidence) D. Raymand, A. Duin, D. Spångberg, W. Goddard, and K. Hermansson, “Water adsorption on stepped ZnO surfaces from MD simulation,” Surface Science. 2010. link Times cited: 135 USED (high confidence) E.-K. Lee and Y.-C. Chung, “Strain-induced Wurtzite to h-BN Phase Transformation in Zinc Oxide nanorods,” 2010 3rd International Nanoelectronics Conference (INEC). 2010. link Times cited: 0 Abstract: Atomistic simulations are utilized to demonstrate a strain-i… read moreAbstract: Atomistic simulations are utilized to demonstrate a strain-induced phase transformation in Zinc Oxide (ZnO) nanowires. The strain-induced transformation occurs by the propagation and annihilation of (0001) planes, and the Wurtzite nanorod is transformed into a hexagonal boron nitride (h-BN) phase. Quantitative structural, mechanical and energetic analyses were performed to verify Wurtzite to h-BN transformation during uniaxial tensile test in [1010] direction. read less USED (low confidence) Y. Gao, Q. Fan, L. Wang, S. Sun, and X. Yu, “Molecular dynamics simulation of oxidation growth of ZnO nanopillars,” Computational Materials Science. 2023. link Times cited: 1 USED (low confidence) N. Sa, S. Chong, H. Q. Wang, and J.-C. Zheng, “Anisotropy Engineering of ZnO Nanoporous Frameworks: A Lattice Dynamics Simulation,” Nanomaterials. 2022. link Times cited: 2 Abstract: The anisotropy engineering of nanoporous zinc oxide (ZnO) fr… read moreAbstract: The anisotropy engineering of nanoporous zinc oxide (ZnO) frameworks has been performed by lattice dynamics simulation. A series of zinc oxide (ZnO) nanoporous framework structures was designed by creating nanopores with different sizes and shapes. We examined the size effects of varying several features of the nanoporous framework (namely, the removal of layers of atoms, surface-area-to-volume ratio, coordination number, porosity, and density) on its mechanical properties (including bulk modulus, Young’s modulus, elastic constant, and Poisson ratio) with both lattice dynamics simulations. We also found that the anisotropy of nanoporous framework can be drastically tuned by changing the shape of nanopores. The maximum anisotropy (defined by Ymax/Ymin) of the Young’s modulus value increases from 1.2 for bulk ZnO to 2.5 for hexagon-prism-shaped ZnO nanoporous framework structures, with a density of 2.72 g/cm3, and, even more remarkably, to 89.8 for a diamond-prism-shape at a density of 1.72 g/cm3. Our findings suggest a new route for desirable anisotropy and mechanical property engineering with nanoporous frameworks by editing the shapes of the nanopores for the desired anisotropy. read less USED (low confidence) K. Sasikumar, R. Ranganathan, S. Rokkam, T. Desai, R. Burnes, and P. G. Cross, “Development of Chemical Kinetics Models from Atomistic Reactive Molecular Dynamics Simulations: Application to Iso-octane Combustion and Rubber Ablative Degradation.,” The journal of physical chemistry. A. 2022. link Times cited: 1 Abstract: Modeling the complex chemical phenomena resulting from multi… read moreAbstract: Modeling the complex chemical phenomena resulting from multiple active species and long-chain polymers is limited by uncertainties in the reaction rate parameters, which increase rapidly with the number of active species and/or reaction pathways. Reactive molecular dynamics simulations have the potential to help obtain in-depth information on the chemical reactions that occur between the polymer (e.g., ablative material) and the multiple active species in an aggressive environment. In this work, we demonstrate that molecular dynamics (MD) simulations using the ReaxFF interatomic potential can be used to obtain the reaction kinetics of complex reaction pathways at high temperatures. We report two recently developed tools, namely, MolfrACT and KinACT, designed to extract chemical kinetic pathways by postprocessing reactive MD simulation data. The pathway extraction is based on a new algorithm, Consistent Reaction Stoichiometry via an Iterative Scheme (CReSIS), for the automated extraction of reactions and kinetics from MD trajectories. As a validation of the methodology, we first report the kinetic analysis and mechanisms for the high-temperature combustion of iso-octane. The observed reaction pathways are consistent with experimental models. In addition, we compare the activation energies of select iso-octane combustion pathways with experimental data and show that nanosecond timescale molecular dynamics simulations are sufficient to obtain realistic estimates of activation energies for different fuel consumption reaction pathways at high temperatures. The framework developed here can potentially be combined with time-series forecasting and machine learning methods to further reduce the computational complexity of transient molecular dynamics simulations, yet yielding realistic chemical kinetics information. Subsequently, the CReSIS scheme applied to ethylene-propylene-diene-monomer (EPDM) rubber ablative reveals that H2O, C2H4, and HCHO are the major products during the initial stages of the polymer degradation in high-temperature oxidative environments. While prior work involving ReaxFF simulations is restricted to overall rates of formation of any species, we extract kinetic information for individual reaction pathways. In this paper, we present several reaction pathways observed during the EPDM rubber degradation into the dominant products and report the pathway-specific reaction rates. Arrhenius analysis reveals that the dominant reaction pathway activation energies for the formation of water, ethylene, and formaldehyde are 34.42, 27.26, and 6.37 kcal/mol, respectively. In contrast, the activation energies for the overall formation (across all reaction pathways) of these products are in the 40-50 kcal/mol range. read less USED (low confidence) H. Jabraoui, S. Gin, T. Charpentier, R. Pollet, and J. Delaye, “Leaching and Reactivity at the Sodium Aluminosilicate Glass–Water Interface: Insights from a ReaxFF Molecular Dynamics Study,” The Journal of Physical Chemistry C. 2021. link Times cited: 13 USED (low confidence) X. Li, L. Liu, H. Mei, S. Xu, J. Li, and J. Zhang, “The formation mechanisms of amorphous bands of boron carbide nanopillars under uniaxial compressions and their effects on mechanical properties from molecular dynamics simulation,” Computational Materials Science. 2021. link Times cited: 1 USED (low confidence) S. Savka, Y. I. Venhryn, A. Serednytski, and D. Popovych, “Molecular dynamics simulation of adsorption processes on the surface of ZnO nanoclusters,” Applied Nanoscience. 2021. link Times cited: 4 USED (low confidence) N. Song, R. Chen, Y. Yang, and X. Li, “Size and Crystal Orientation-Dependent Thermal Behaviors of ZnO Nanobelts,” Journal of Physical Chemistry C. 2020. link Times cited: 2 USED (low confidence) R. Dongol, L. Wang, A. Cormack, and S. Sundaram, “Molecular dynamics simulation of sodium aluminosilicate glass structures and glass surface-water reactions using the reactive force field (ReaxFF),” Applied Surface Science. 2018. link Times cited: 36 USED (low confidence) S.-E. Zhang, F. Cheng, X. He, and Z. Chen, “ReaxFF Molecular Dynamic Simulations of ZnO Nanocluster and Films in H2 Atmosphere,” Journal of Physical Chemistry C. 2017. link Times cited: 0 Abstract: Reactive molecular dynamics simulations were performed to ex… read moreAbstract: Reactive molecular dynamics simulations were performed to explore the structural evolution of a ZnO nanocluster and (0001) and (1010) surfaces under a H2 atmosphere at different temperatures. The mechanisms of H2 dissociation and water formation were analyzed. Our simulations reveal that there are two pathways for H2 dissociation and three routes for water formation on the surfaces. The nanocluster is more active for H2 dissociation and water formation than the two surfaces. The gas–solid interactions lead to outward displacement of the substrate O atoms. While the O-terminated surface of the (0001) facet is active for H2 dissociation and water formation, the Zn-terminated one is inactive for the dissociation. Unlike the (0001) surface which is more easily reduced, the (1010) surface is readily hydroxylated. Water formation and desorption results in surface oxygen depletion and Zn aggregation which lead to surface metallization, in accordance with the experimental observations. Our simulations show that... read less USED (low confidence) N. Wang, J.-hua Peng, A.-min Pang, T. He, F. Du, and A. Jaramillo-Botero, “Thermodynamic Simulation of the RDX–Aluminum Interface Using ReaxFF Molecular Dynamics,” Journal of Physical Chemistry C. 2017. link Times cited: 39 Abstract: We use reactive molecular dynamics (RMD) simulations to stud… read moreAbstract: We use reactive molecular dynamics (RMD) simulations to study the interface between cyclotrimethylene trinitramine (RDX) and aluminum (Al) with different oxide layers to elucidate the effect of nanosized Al on thermal decomposition of RDX. A published ReaxFF force field for C/H/N/O elements was retrained to incorporate Al interactions and then used in RMD simulations to characterize compound energetic materials. We find that the predicted adsorption energies for RDX on the Al(111) surface and the apparent activation energies of RDX and RDX/Al are in agreement with ab initio calculations. The Al(111) surface-assisted decomposition of RDX occurs spontaneously without potential barriers, but the decomposition rate becomes slow when compared with that for RDX powder. We also find that the Al(111) surface with an oxide layer (Al oxide) slightly increases the potential barriers for decomposition of RDX molecules, while α-Al2O3(0001) retards thermal decomposition of RDX, due to the changes in thermal decompositi... read less USED (low confidence) Y. Liu, C. Dong, B. Z. Sun, and Y. Qi, “Effect of temperature on the growth of a-axis ZnO films: a reactive force field-based molecular dynamics study,” Journal of Materials Research. 2017. link Times cited: 0 Abstract: Understanding film initiation and growth mechanisms at the a… read moreAbstract: Understanding film initiation and growth mechanisms at the atomic level is crucial to obtain high-quality nonpolar ZnO films. Using the advanced reactive force field-based molecular dynamics method, we theoretically studied the effect of substrate temperature (350–950 K) on the quality, layer develop mechanism and defect formation of ZnO films. Investigation of the energy, radial distribution function, layer coverage, sputtering and injecting phenomena indicated that the present films grown at 500–600 K possessed the optimal quality. Further investigation of the growth condition, instant film profiles, interfacial microstructure evolutions and layered snapshots revealed that, addition of atoms on newly formed localized films can induce some partially bonded or extruded atoms out of the film plane. Further adherence of depositing atoms to these unstable or extruded atoms induces the initiation and growth of a new layer. read less USED (low confidence) S. Savka, D. Popovych, and A. Serednytski, “Molecular Dynamics Simulations of the Formation Processes of Zinc Oxide Nanoclusters in Oxygen Environment.” 2016. link Times cited: 10 USED (low confidence) Y. Liu, M. Shahzad, and Y. Qi, “Growth of a-axis ZnO films on the defective substrate with different O/Zn ratios: A reactive force field based molecular dynamics study,” Journal of Alloys and Compounds. 2015. link Times cited: 7 USED (low confidence) C. Zou, Y. Shin, A. V. van Duin, H. Fang, and Z.-kui Liu, “Molecular dynamics simulations of the effects of vacancies on nickel self-diffusion, oxygen diffusion and oxidation initiation in nickel, using the ReaxFF reactive force field,” Acta Materialia. 2015. link Times cited: 76 USED (low confidence) I. Schäfer, G. Lasko, T. Do, J. Pleiss, U. Weber, and S. Schmauder, “Peptide–zinc oxide interaction: Finite element simulation using cohesive zone models based on molecular dynamics simulation,” Computational Materials Science. 2014. link Times cited: 4 USED (low confidence) Y.-T. Cheng et al., “Atomistic simulations of the adsorption and migration barriers of Cu adatoms on ZnO surfaces using COMB potentials,” Surface Science. 2012. link Times cited: 25 USED (low confidence) A. Bharathi, A. M. Kamat, and A. Duin, “Study of effect of water vapor and mechanical strain on thermal conductivity of zinc oxide using the ReaxFF reactive force field,” Computational and Theoretical Chemistry. 2012. link Times cited: 11 USED (low confidence) B. Devine et al., “Atomistic simulations of copper oxidation and Cu/Cu2O interfaces using charge-optimized many-body potentials,” Physical Review B. 2011. link Times cited: 64 Abstract: Bryce Devine,1 Tzu-Ray Shan( ),1 Yu-Ting Cheng( ),1 Alan J. … read moreAbstract: Bryce Devine,1 Tzu-Ray Shan( ),1 Yu-Ting Cheng( ),1 Alan J. H. McGaughey,1,2 Minyoung Lee,2 Simon R. Phillpot,1 and Susan B. Sinnott1,* 1Department of Materials Science and Engineering, University of Florida, Gainesville, FL 32611-6400, USA 2Department of Mechanical Engineering, Carnegie-Mellon University, Pittsburgh, PA, 15213, USA (Received 14 February 2011; revised manuscript received 21 July 2011; published 12 September 2011) read less USED (low confidence) S. Savka, I. Mohylyak, and D. Popovych, “Influence of defects on adsorption processes in the near-surface layers of ZnO nanoclusters: MD study,” Journal of Physical Studies. 2022. link Times cited: 1 USED (low confidence) S. Kawagishi et al., “Effect of Polarity of a Substrate on ZnO Crystal Growth Process by Molecular Dynamics Simulation,” Journal of Computer Chemistry, Japan. 2017. link Times cited: 0 NOT USED (low confidence) M. Nyepetsi, O. Oyetunji, and F. Mbaiwa, “ReaxFF study of the decarboxylation of methyl palmitate over binary metallic nickel-molybdenum catalysts,” Molecular Simulation. 2023. link Times cited: 0 NOT USED (low confidence) M. Saeedimasine, F. Grote, and A. Lyubartsev, “Ab Initio Derived Classical Force Field for Molecular Dynamics Simulations of ZnO Surfaces in Biological Environment,” The Journal of Physical Chemistry. a. 2023. link Times cited: 0 Abstract: Zinc oxide nanostructures are used in an ever increasing lin… read moreAbstract: Zinc oxide nanostructures are used in an ever increasing line of applications in technology and biomedical fields. This requires a detailed understanding of the phenomena that occur at the surface particularly in aqueous environments and in contact with biomolecules. In this work, we used ab initio molecular dynamics (AIMD) simulations to determine structural details of ZnO surfaces in water and to develop a general and transferable classical force field for hydrated ZnO surfaces. AIMD simulations show that water molecules dissociate near unmodified ZnO surfaces, forming hydroxyl groups at about 65% of the surface Zn atoms and protonating 3-coordinated surface oxygen atoms, while the rest of the surface Zn atoms bind molecularly adsorbed waters. Several force field atom types for ZnO surface atoms were identified by analysis of the specific connectivities of atoms. The analysis of the electron density was then used to determine partial charges and Lennard-Jones parameters for the identified force field atom types. The obtained force field was validated by comparison with AIMD results and with available experimental data on adsorption and immersion enthalpies, as well as adsorption free energies of several amino acids in methanol. The developed force field can be used for modeling of ZnO in aqueous and other fluid environments and in interaction with biomolecules. read less NOT USED (low confidence) L. He, Z. Cui, X. Sun, J. Zhao, and D. Wen, “Sensitivity Analysis of the Catalysis Recombination Mechanism on Nanoscale Silica Surfaces,” Nanomaterials. 2022. link Times cited: 2 Abstract: A deep understanding of surface catalysis recombination char… read moreAbstract: A deep understanding of surface catalysis recombination characteristics is significant for accurately predicting the aeroheating between hypersonic non-equilibrium flow and thermal protection materials, while a de-coupling sensitivity analysis of various influential factors is still lacking. A gas–solid interface (GSI) model with a hyperthermal flux boundary was established to investigate the surface catalysis recombination mechanisms on nanoscale silica surfaces. Using the reactive molecular dynamics (RMD) simulation method, the effects of solid surface temperature, gas incident angle, and translational energy on the silica surface catalysis recombination were qualified under hyperthermal atomic oxygen (AO), atomic nitrogen (AN), and various AN/AO gas mixtures’ influence. It can be found that, though the Eley–Rideal (E–R) recombination mechanism plays a dominant role over the Langmuir–Hinshelwood (L–H) mechanism for all the sensitivity analyses, a non-linear increasing pattern of AO recombination coefficient γO2 with the increase in incident angle θin and translational energy Ek is observed. Compared with the surface catalysis under hyperthermal AO impact, the AN surface adsorption fraction shows an inverse trend with the increase in surface temperature, which suggests the potential inadequacy of the traditional proportional relationship assumptions between the surface adsorption concentration and the surface catalysis recombination coefficient for other species’ impact instead of AOs. For the incoming bi-component AO/AN gas mixtures, the corresponding surface catalysis coefficient is not the simple superposition of the effects of individual gases but is affected by both the intramolecular bond energies (e.g., O2, N2) and intermolecular energies (e.g., Si/N, Si/O). read less NOT USED (low confidence) X.-W. Wang, X. Sun, T. Song, J. Tian, and Z.-J. Liu, “Development of the new interatomic potentials for the wurtzite phase of ZnO,” Applied Physics A. 2022. link Times cited: 1 NOT USED (low confidence) I. Brown, R. Smith, and S. Kenny, “A ReaxFF potential for Al–ZnO systems,” Modelling and Simulation in Materials Science and Engineering. 2022. link Times cited: 0 Abstract: A reactive field force potential has been created in order t… read moreAbstract: A reactive field force potential has been created in order to model the structural effects of low percentage dopant aluminium in a zinc oxide (ZnO) system. The potential’s parameters were fitted to configurations computed with density functional theory: binding energies were considered for surface structures and for Al in ZnO bulk crystals. Energies for Zn–Al alloys were also considered. Forces were fit to zero for all equilibrium structures and were also fitted for some non-equilibrium structures. As a first application of the model, the energetic deposition (0.1–40 eV) of an aluminium atom onto the polar surface of a ZnO (0001¯) is considered. For low energies the Al atom attaches to two preferred sites on the surface but as the energy increases above ≈15 eV subplantation is preferred at near normal incidence, with high diffusion barriers between stable sites. At these energies, reflection of the Al atom occurs at incident angles above ≈55° . read less NOT USED (low confidence) T. Nie and X.-qing Gong, “A theoretical study of the twinned ZnO nanostructures,” Applied Surface Science. 2022. link Times cited: 1 NOT USED (low confidence) Y. Zhang, R. Chen, J. Riviere, and J. Comer, “Extraction of Chlorobenzenes and PCBs from Water by ZnO Nanoparticles,” Processes. 2021. link Times cited: 0 Abstract: Metal oxide nanoparticles have great potential for selective… read moreAbstract: Metal oxide nanoparticles have great potential for selective adsorption and catalytic degradation of contaminants from aqueous solutions. In this study, we employ mass spectrometry and molecular dynamics simulations to better understand the chemical and physical mechanisms determining the affinity of chlorobenzenes and polychlorinated biphenyls (PCBs) for zinc oxide nanoparticles (ZnO NPs). The experiments and simulations both demonstrate that the adsorption coefficients for chlorobenzenes increase steadily with the number of chlorine atoms, while, for PCBs, the relation is more complex. The simulations link this complexity to chlorine atoms at ortho positions hindering coplanar conformations. For a given number of chlorine atoms, the simulations predict decreasing adsorption affinity with increasing numbers of ortho substitutions. Consequently, the simulations predict that some of the highest adsorption affinities for ZnO NPs are exhibited by dioxin-like PCBs, suggesting the possibility of selective sequestration of these most acutely toxic PCBs. Remarkably, the experiments show that the PCB adsorption coefficients of ZnO NPs with diameters ≤ 80 nm exceed those of a soil sample by 5–7 orders of magnitude, meaning that a single gram of ZnO NPs could sequester low levels of PCB contamination from as much as a ton of soil. read less NOT USED (low confidence) K. McKenna, “Atomistic Modeling of Oxide Nanoparticles,” Metal Oxide Nanoparticles. 2021. link Times cited: 0 NOT USED (low confidence) R. A. Gallivan and J. Greer, “Failure Mechanisms in Vertically Aligned Dense Nanowire Arrays.,” Nano letters. 2021. link Times cited: 1 Abstract: Nanowires are an increasingly prevalent class of nanomateria… read moreAbstract: Nanowires are an increasingly prevalent class of nanomaterials in composites and devices, with arrays and other complex geometries used in various applications. Little investigation has been done regarding the mechanical behavior of micron-sized nanowire structures. We conduct in situ microcompression experiments on vertically aligned dense microbundles of 300 nm diameter single-crystalline zinc oxide nanowires to gain insights into their structural failure. Experiments demonstrate that bundles containing approximately 10-130 nanowires experience two failure regimes: (1) localized noncatastrophic interfacial splitting and (2) global structural failure. Utilizing Weibull statistics and experimental results, we develop a technique for analyzing flaw distribution and use it to predict the expected range of bundle failure stress. This analysis provides guidelines for nanowire arrays' susceptibility to failure, sensitivity to flaw size, interfacial interactions of constituents, and degree of alignment. This work develops insights to understand and predict fundamental failure mechanisms in highly aligned, dense structures. read less NOT USED (low confidence) C. M. Daksha, J. Yeon, S. Chowdhury, and J. Gillespie, “Automated ReaxFF parametrization using machine learning,” Computational Materials Science. 2021. link Times cited: 19 NOT USED (low confidence) R. Wang, C. Wang, Y. Feng, and C. Tang, “Mechanistic pathway of water adsorption to impact on the nonlinear elasticity of single-crystalline ZnO NWs,” Computational Materials Science. 2020. link Times cited: 0 NOT USED (low confidence) A. Rahnamoun et al., “ReaxFF/AMBER-A Framework for Hybrid Reactive/Nonreactive Force Field Molecular Dynamics Simulations.,” Journal of chemical theory and computation. 2020. link Times cited: 15 Abstract: Combined quantum mechanical/molecular mechanical (QM/MM) mod… read moreAbstract: Combined quantum mechanical/molecular mechanical (QM/MM) models using semiempirical and ab initio methods have been extensively reported on over the past few decades. These methods have been shown to be capable of providing unique insights into a range of problems, but they are still limited to relatively short time scales, especially QM/MM models using ab initio methods. An intermediate approach between a QM based model and classical mechanics could help fill this time-scale gap and facilitate the study of a range of interesting problems. Reactive force fields represent the intermediate approach explored in this paper. A widely used reactive model is ReaxFF, which has largely been applied to materials science problems and is generally used as a stand-alone (i.e., the full system is modeled using ReaxFF). We report a hybrid ReaxFF/AMBER molecular dynamics (MD) tool, which introduces ReaxFF capabilities to capture bond breaking and formation within the AMBER MD software package. This tool enables us to study local reactive events in large systems at a fraction of the computational costs of QM/MM models. We describe the implementation of ReaxFF/AMBER, validate this implementation using a benzene molecule solvated in water, and compare its performance against a range of similar approaches. To illustrate the predictive capabilities of ReaxFF/AMBER, we carried out a Claisen rearrangement study in aqueous solution. In a first for ReaxFF, we were able to use AMBER's potential of mean force (PMF) capabilities to perform a PMF study on this organic reaction. The ability to capture local reaction events in large systems using combined ReaxFF/AMBER opens up a range of problems that can be tackled using this model to address both chemical and biological processes. read less NOT USED (low confidence) L. G. Lascane, E. Oliveira, D. Galvão, and A. Batagin‐Neto, “Polyfuran-based chemical sensors: Identification of promising derivatives via DFT calculations and fully atomistic reactive molecular dynamics,” European Polymer Journal. 2020. link Times cited: 15 NOT USED (low confidence) C. Zhao et al., “Precipitation of calcium-alumino-silicate-hydrate gels: The role of the internal stress.,” The Journal of chemical physics. 2020. link Times cited: 9 Abstract: Concrete gains its strength from the precipitation of a calc… read moreAbstract: Concrete gains its strength from the precipitation of a calcium-alumino-silicate-hydrate (C-A-S-H) colloidal gel, which acts as its binding phase. However, despite concrete's ubiquity in the building environment, the atomic-scale mechanism of C-A-S-H precipitation is still unclear. Here, we use reactive molecular dynamics simulations to model the early-age precipitation of a C-A-S-H gel. We find that, upon gelation, silicate and aluminate precursors condensate and polymerize to form an aluminosilicate gel network. Notably, we demonstrate that the gelation reaction is driven by the existence of a mismatch of atomic-level internal stress between Si and Al polytopes, which are initially experiencing some local tension and compression, respectively. The polymerization of Si and Al polytopes enables the release of these competitive stresses. read less NOT USED (low confidence) Z. Liu, T. Yunqing, N. Liao, and P. Yang, “Study on interfacial interaction between Si and ZnO,” Ceramics International. 2019. link Times cited: 15 NOT USED (low confidence) Y. I. Venhryn, S. Savka, R. Bovhyra, V. M. Zhyrovetsky, A. Serednytski, and D. Popovych, “Obtaining, structure and gas sensor properties of nanopowder metal oxides,” Materials Today: Proceedings. 2019. link Times cited: 9 NOT USED (low confidence) R. DeLong, J. Comer, E. Mathew, and M. Jaberi-Douraki, “Comparative Molecular Immunological Activity of Physiological Metal Oxide Nanoparticle and its Anticancer Peptide and RNA Complexes,” Nanomaterials. 2019. link Times cited: 13 Abstract: Currently, there is a great interest in nanoparticle-based v… read moreAbstract: Currently, there is a great interest in nanoparticle-based vaccine delivery. Recent studies suggest that nanoparticles when introduced into the biological milieu are not simply passive carriers but may also contribute immunological activity themselves or of their own accord. For example there is considerable interest in the biomedical applications of one of the physiologically-based inorganic metal oxide nanoparticle, zinc oxide (ZnO). Indeed zinc oxide (ZnO) NP are now recognized as a nanoscale chemotherapeutic or anticancer nanoparticle (ANP) and several recent reports suggest ZnO NP and/or its complexes with drug and RNA induce a potent antitumor response in immuno-competent mouse models. A variety of cell culture studies have shown that ZnO NP can induce cytokines such as IFN-γ, TNF-α, IL-2, and IL-12 which are known to regulate the tumor microenvironment. Much less work has been done on magnesium oxide (MgO), cobalt oxide (Co3O4), or nickel oxide (NiO); however, despite the fact that these physiologically-based metal oxide NP are reported to functionally load and assemble RNA and protein onto their surface and may thus also be of potential interest as nanovaccine platform. Here we initially compared in vitro immunogenicity of ZnO and Co3O4 NP and their effects on cancer-associated or tolerogenic cytokines. Based on these data we moved ZnO NP forward to testing in the ex vivo splenocyte assay relative to MgO and NiO NP and these data showed significant difference for flow cytometry sorted population for ZnO-NP, relative to NiO and MgO. These data suggesting both molecular and cellular immunogenic activity, a double-stranded anticancer RNA (ACR), polyinosinic:poly cytidylic acid (poly I:C) known to bind ZnO NP; when ZnO-poly I:C was injected into B16F10-BALB/C tumor significantly induced, IL-2 and IL-12 as shown by Cohen’s d test. LL37 is an anticancer peptide (ACP) currently in clinical trials as an intratumoral immuno-therapeutic agent against metastatic melanoma. LL37 is known to bind poly I:C where it is thought to compete for receptor binding on the surface of some immune cells, metastatic melanoma and lung cells. Molecular dynamic simulations revealed association of LL37 onto ZnO NP confirmed by gel shift assay. Thus using the well-characterized model human lung cancer model cell line (BEAS-2B), poly I:C RNA, LL37 peptide, or LL37-poly I:C complexes were loaded onto ZnO NP and delivered to BEAS-2B lung cells, and the effect on the main cancer regulating cytokine, IL-6 determined by ELISA. Surprisingly ZnO-LL37, but not ZnO-poly I:C or the more novel tricomplex (ZnO-LL37-poly I:C) significantly suppressed IL-6 by >98–99%. These data support the further evaluation of physiological metal oxide compositions, so-called physiometacomposite (PMC) materials and their formulation with anticancer peptide (ACP) and/or anticancer RNA (ACR) as a potential new class of immuno-therapeutic against melanoma and potentially lung carcinoma or other cancers. read less NOT USED (low confidence) G. Barcaro, S. Monti, L. Sementa, and V. Carravetta, “Modeling Nucleation and Growth of ZnO Nanoparticles in a Low Temperature Plasma by Reactive Dynamics.,” Journal of chemical theory and computation. 2019. link Times cited: 5 Abstract: The very first stages of nucleation and growth of ZnO nanopa… read moreAbstract: The very first stages of nucleation and growth of ZnO nanoparticles in a plasma reactor are studied by means of a multiscale computational paradigm where the DFT-GGA approach is used to evaluate structure and electronic energy of small (ZnO) N clusters ( N ≤ 24) that are employed as a training set (TS) for the optimization of a Reactive Force Field (ReaxFF). Reactive Molecular Dynamics (RMD) simulations based on this tuned ReaxFF are carried out to reproduce nucleation and growth in a realistic environment. Inside the reaction chamber the temperature is around 1200 K, and the zinc atoms are oxidized in an oxygen-rich atmosphere at high pressure (about 20 atm), whereas in the quenching chamber where the temperature is lower (about 800 K) the ZnO embryo-nanoclusters are grown. The main processes ruling gas-phase nucleation and growth of ZnO nanoclusters are identified and discussed together with the dependence of the inception time and average stoichiometry of nanoclusters of different size on the composition of precursor material and physical parameters. read less NOT USED (low confidence) S. Thinius and T. Bredow, “Spectroscopic Properties of Chalcopyrite Nanoparticles,” The Journal of Physical Chemistry C. 2019. link Times cited: 2 Abstract: An approach is presented to construct stoichiometric and sat… read moreAbstract: An approach is presented to construct stoichiometric and saturated clusters as representative models for nanoparticles based on quantum-chemical surface energy calculations. The procedure consists of three steps. In the first step, the shape of the cluster is determined by applying the Wulff theorem based on calculated surface energies of the solid compound. Stoichiometry is recovered by adding selected atoms on the cluster surface. If the particles in solution are to be modeled, solvent molecules may be added. A global optimization is then performed to allow for full reconstruction of the cluster structure. As an example, we studied the spectroscopic properties of chalcopyrite (CuFeS2) nanoparticles and compared our calculated results to experimental O 1s X-ray photoelectron spectroscopy, IR, and optical spectra. read less NOT USED (low confidence) M. Sengul, C. Randall, and A. V. van Duin, “ReaxFF Molecular Dynamics Study on the Influence of Temperature on Adsorption, Desorption, and Decomposition at the Acetic Acid/Water/ZnO(101̅0) Interface Enabling Cold Sintering.,” ACS applied materials & interfaces. 2018. link Times cited: 33 Abstract: The reaction dynamics of a liquid-solid interface with the e… read moreAbstract: The reaction dynamics of a liquid-solid interface with the example of an acetic acid/water solution interacting with a ZnO(101̅0) surface was investigated using ReaxFF reactive force field-based molecular dynamics. The interactions were studied over a broad temperature range to assess the kinetics and reaction pathways. Two different acetic acid dissociation mechanisms are observed in the simulations: (1) deprotonation to surface cation, which produces a terminal hydroxyl and (2) deprotonation to a bridging hydroxyl, which results in water production. An increase in temperature promotes the dissociation of acetic acids and its adsorption to surface at first, but as the temperature increase continues, the surface coverage by acetates decreases due to evaporation from the surface or decomposition. The acetate decomposition starts with a nucleophilic attack of oxygen to methyl carbon and results in the production of carbon dioxide, which is consistent with experimental findings in the literature. The coverage of the surface by water molecules decreases as the system is heated up, which is also observed in other molecular dynamics studies. At elevated temperatures, acetate molecules are more stable than water molecules or bridging hydroxyls on the surface. These simulations validate the ReaxFF method for the water/organic mixture and metal oxide surface interactions and provide insights into structure and reactivity of aqueous solvents on metal oxide surfaces at elevated temperatures. Adsorption trends that are observed in this study are consistent with phenomenological Langmuir models. The reaction of acetic acid decomposition to smaller molecules such as CO2 and CH2O agrees with experimental observations. Understanding the details of these dynamic surface reactions are critical to understand important new cold sintering processes that utilize transient liquid and solid reactions, and the latter could be used to predict solvent selection for cold sintering. read less NOT USED (low confidence) H.-S. Jang, K.-M. Kim, and B.-J. Lee, “Modified embedded-atom method interatomic potentials for pure Zn and Mg-Zn binary system,” Calphad-computer Coupling of Phase Diagrams and Thermochemistry. 2018. link Times cited: 23 NOT USED (low confidence) F. Kong et al., “Charge-transfer modified embedded atom method dynamic charge potential for Li–Co–O system,” Journal of Physics: Condensed Matter. 2017. link Times cited: 4 Abstract: To overcome the limitation of conventional fixed charge pote… read moreAbstract: To overcome the limitation of conventional fixed charge potential methods for the study of Li-ion battery cathode materials, a dynamic charge potential method, charge-transfer modified embedded atom method (CT-MEAM), has been developed and applied to the Li–Co–O ternary system. The accuracy of the potential has been tested and validated by reproducing a variety of structural and electrochemical properties of LiCoO2. A detailed analysis on the local charge distribution confirmed the capability of this potential for dynamic charge modeling. The transferability of the potential is also demonstrated by its reliability in describing Li-rich Li2CoO2 and Li-deficient LiCo2O4 compounds, including their phase stability, equilibrium volume, charge states and cathode voltages. These results demonstrate that the CT-MEAM dynamic charge potential could help to overcome the challenge of modeling complex ternary transition metal oxides. This work can promote molecular dynamics studies of Li ion cathode materials and other important transition metal oxides systems that involve complex electrochemical and catalytic reactions. read less NOT USED (low confidence) C. Jacobs et al., “UV-activated ZnO films on a flexible substrate for room temperature O2 and H2O sensing,” Scientific Reports. 2017. link Times cited: 57 NOT USED (low confidence) M. Molinari, A. Brukhno, S. C. Parker, and D. Spagnoli, “Force Field Application and Development.” 2016. link Times cited: 1 NOT USED (low confidence) J. Wang et al., “Ligand-directed rapid formation of ultralong ZnO nanowires by oriented attachment for UV photodetectors,” Journal of Materials Chemistry C. 2016. link Times cited: 22 Abstract: Ultralong ZnO nanowires with lengths of 20–80 μm and aspect … read moreAbstract: Ultralong ZnO nanowires with lengths of 20–80 μm and aspect ratios of 200–500 were synthesized within 15 minutes via a low-temperature hydrothermal method. With the assistance of sodium dodecyl sulfonate (SDSN) as the capping ligand, ZnO nanowires were formed by the initial nucleation of nanocrystals followed by the ligand-directed oriented attachment. Head-to-head attachment, side-by-side coalescence and nanocrystals attached to the surfaces were observed at different growth stages. ZnO microrods (lengths: 2–10 μm, diameters: 0.5 to 5 μm) were formed in the absence of SDSN. FT-IR spectra, XPS analysis and molecular dynamics simulations revealed that SDSN molecules were preferentially adsorbed onto the (100) planes rather than polar (001) planes, with their sulfonate groups coordinating with the surface zinc ions and possibly forming Zn–SO3 complexes. Such selective adsorption not only protected the initially nucleated ZnO nanocrystals from rapid aggregation, but also directed their subsequent self-assembly into highly-anisotropic nanowires. The as-prepared ZnO nanowires exhibited improved photoluminescence properties compared to the microrods. I–V characteristics indicated that the ZnO nanowires exhibited a much lower dark current, while an enhanced photocurrent upon 360 nm light illumination compared to the microrods. In addition, UV photodetectors using ZnO nanowires showed 27 times higher photo-sensitivity and 15.4/13.8 times higher rise/decay rates compared to those using ZnO microrods, which were attributed to the morphological effects in addition to the improved optical properties. read less NOT USED (low confidence) T. Senftle et al., “The ReaxFF reactive force-field: development, applications and future directions.” 2016. link Times cited: 1212 NOT USED (low confidence) H. Li, P. Winget, and J. Brédas, “Transparent Conducting Oxides of Relevance to Organic Electronics: Electronic Structures of Their Interfaces with Organic Layers,” Chemistry of Materials. 2014. link Times cited: 46 Abstract: Transparent conducting oxides (TCO) are a critical component… read moreAbstract: Transparent conducting oxides (TCO) are a critical component of many organic electronic devices including organic solar cells and light-emitting diodes. In this Perspective, we discuss what we have learned from our theoretical investigations, at the density functional theory (DFT) level, of the electronic structures of several technologically relevant transparent conducting oxides and their interfaces with organic layers. In particular, we describe how DFT calculations can be used to provide a detailed understanding of (i) the impact of surface modification by an organic monolayer on the interfacial electronic structure and the work function; (ii) the electronic characteristics of TCO surfaces as a function of surface hydroxylation and the presence of various intrinsic and extrinsic defects; and (iii) the nature of the charge transfer taking place between an organic semiconducting layer and a TCO electrode when considering the physisorption of a monolayer of π-conjugated organic molecules on the TCO surfaces. read less NOT USED (low confidence) H. Hu, H. Ji, and Y. Sun, “The effect of oxygen vacancies on water wettability of a ZnO surface.,” Physical chemistry chemical physics : PCCP. 2013. link Times cited: 85 Abstract: In this study, the effect of oxygen vacancies on the water w… read moreAbstract: In this study, the effect of oxygen vacancies on the water wettability of a hydrated ZnO(100) surface has been examined via molecular dynamics simulations with a reactive force field (ReaxFF). The results show that the oxygen vacancies on the ZnO surface change the structures of the ZnO surface and subsequently its water adsorption capability. While a 1 : 1 ratio of water to hydroxyl is observed for a water monolayer absorbed on ZnO(100) without oxygen vacancies, additional water adsorption as coordinate hydroxyl that resides on the vacancy site and bonds with three lattice zinc atoms is observed on the surfaces with oxygen vacancies. The results also show that the energy of the interaction per unit area between water and the hydrated ZnO surface is 55.1% higher in the presence of the oxygen vacancies than that without oxygen vacancies. This leads to a water contact angle of ~115° for the hydrated ZnO(100) surface in the absence of vacancies and ~21° with vacancies. The wetting kinetics of a water droplet on a ZnO(100) surface with and without oxygen vacancies are compared with the diffusion-limited reactive wetting and molecular kinetics models, respectively. In addition, the ordering of the vacancy sites is found not to significantly affect the wettability of the ZnO(100) surface. read less NOT USED (low confidence) T. Liang et al., “Classical atomistic simulations of surfaces and heterogeneous interfaces with the charge-optimized many body (COMB) potentials,” Materials Science & Engineering R-reports. 2013. link Times cited: 207 NOT USED (low confidence) G. Nawrocki and M. Cieplak, “Amino acids and proteins at ZnO-water interfaces in molecular dynamics simulations.,” Physical chemistry chemical physics : PCCP. 2013. link Times cited: 36 Abstract: We determine potentials of the mean force for interactions o… read moreAbstract: We determine potentials of the mean force for interactions of amino acids with four common surfaces of ZnO in aqueous solutions. The method involves all-atom molecular dynamics simulations combined with the umbrella sampling technique. The profiled nature of the density of water with the strongly adsorbed first layer affects the approach of amino acids to the surface and generates either repulsion or weak binding. The largest binding energy is found for tyrosine interacting with the surface in which the Zn ions are at the top. It is equal to 7 kJ mol(-1) which is comparable to that of the hydrogen bonds in a protein. This makes the adsorption of amino acids onto the ZnO surface much weaker than onto the well studied surface of gold. Under vacuum, binding energies are more than 40 times stronger (for one of the surfaces). The precise manner in which water molecules interact with a given surface influences the binding energies in a way that depends on the surface. Among the four considered surfaces the one with Zn at the top is recognized as binding almost all amino acids with an average binding energy of 2.60 kJ mol(-1). Another (O at the top) is non-binding for most amino acids. For binding situations the average energy is 0.66 kJ mol(-1). The remaining two surfaces bind nearly as many amino acids as they do not and the average binding energies are 1.46 and 1.22 kJ mol(-1). For all of the surfaces the binding energies vary between amino acids significantly: the dispersion in the range of 68-154% of the mean. A small protein is shown to adsorb onto ZnO only intermittently and with only a small deformation. Various adsorption events lead to different patterns in mobilities of amino acids within the protein. read less NOT USED (low confidence) A. Asthana and D. Wheeler, “A polarizable reactive force field for water to enable molecular dynamics simulations of proton transport.,” The Journal of chemical physics. 2013. link Times cited: 6 Abstract: A new polarizable water model is developed for molecular dyn… read moreAbstract: A new polarizable water model is developed for molecular dynamics (MD) simulations of the proton transport process. The interatomic potential model has three important submodels corresponding to electrostatic interactions, making and breaking of covalent bonds, and treatment of electron exchange and correlation through a van der Waals potential. A polarizable diffuse charge density function was used to describe Coulombic interactions between atoms. Most of the model parameters were obtained from ab initio data for a lone water molecule. Molecules respond realistically to their electrochemical environment by the use of coupled fluctuating charge and fluctuating dipole dynamics, which controlled the charge density. The main purpose of the work is to develop a general model and framework for future studies, though some validation work was performed here. We applied the model to a MD simulation study of bulk properties of liquid water at room temperature and model gave good agreement with thermodynamic and transport properties at the same conditions. The model was then applied to a preliminary study of proton transfer, in which multiple proton transfer events were observed, though the rate of proton transfer was under-predicted by a factor of 5. read less NOT USED (low confidence) A. Ahmad, A. A. Abdulkarim, B. .. Ooi, and S. Ismail, “Recent development in additives modifications of polyethersulfone membrane for flux enhancement,” Chemical Engineering Journal. 2013. link Times cited: 284 NOT USED (low confidence) B. D. Jensen, A. Bandyopadhyay, K. Wise, and G. Odegard, “Parametric Study of ReaxFF Simulation Parameters for Molecular Dynamics Modeling of Reactive Carbon Gases.,” Journal of chemical theory and computation. 2012. link Times cited: 35 Abstract: The development of innovative carbon-based materials can be … read moreAbstract: The development of innovative carbon-based materials can be greatly facilitated by molecular modeling techniques. Although the Reax Force Field (ReaxFF) can be used to simulate the chemical behavior of carbon-based systems, the simulation settings required for accurate predictions have not been fully explored. Using the ReaxFF, molecular dynamics (MD) simulations are used to simulate the chemical behavior of pure carbon and hydrocarbon reactive gases that are involved in the formation of carbon structures such as graphite, buckyballs, amorphous carbon, and carbon nanotubes. It is determined that the maximum simulation time step that can be used in MD simulations with the ReaxFF is dependent on the simulated temperature and selected parameter set, as are the predicted reaction rates. It is also determined that different carbon-based reactive gases react at different rates, and that the predicted equilibrium structures are generally the same for the different ReaxFF parameter sets, except in the case of the predicted formation of large graphitic structures with the Chenoweth parameter set under specific conditions. read less NOT USED (low confidence) D. A. Newsome, D. Sengupta, H. Foroutan, M. F. Russo, and A. Duin, “Oxidation of Silicon Carbide by O2 and H2O: A ReaxFF Reactive Molecular Dynamics Study, Part I,” Journal of Physical Chemistry C. 2012. link Times cited: 158 Abstract: Simulations of the initial oxidation process of a SiC surfac… read moreAbstract: Simulations of the initial oxidation process of a SiC surface exposed to O2 and H2O molecules was studied with ReaxFF, an atomically detailed reactive molecular dynamics method that naturally models the breaking and forming of bonds. In this work, the ReaxFF forcefield was first expanded by training it with new quantum mechanics data of the binding energy, equation of state, and heat of formation of the SiC crystal, along with data from earlier studies that describes Si – Si, Si – O, and Si – H interactions. This expanded ReaxFF forcefield is capable of simultaneously describing both Si–C–O and Si–O–H bonding interactions. Using the forcefield, oxidation simulations were performed at various temperatures (in the range of 500 to 5000 K), and the trajectories were analyzed. The analyses showed that SiC gradually transforms into the oxides of silicon with simultaneous formation of a graphite-like layer. In presence of excess O2, the graphite-like layer was further oxidized to CO and CO2. We also analyzed the... read less NOT USED (low confidence) D. Raymand, T. Jacobsson, K. Hermansson, and T. Edvinsson, “Investigation of Vibrational Modes and Phonon Density of States in ZnO Quantum Dots,” Journal of Physical Chemistry C. 2012. link Times cited: 40 Abstract: The ability to understand the phonon behavior in small metal… read moreAbstract: The ability to understand the phonon behavior in small metal oxide nanostructures and their surfaces is of great importance for thermal and microelectronic applications in successively smaller devi ... read less NOT USED (low confidence) D. Bedrov, G. D. Smith, and A. V. van Duin, “Reactions of singly-reduced ethylene carbonate in lithium battery electrolytes: a molecular dynamics simulation study using the ReaxFF.,” The journal of physical chemistry. A. 2012. link Times cited: 130 Abstract: We have conducted quantum chemistry calculations and gas- an… read moreAbstract: We have conducted quantum chemistry calculations and gas- and solution-phase reactive molecular dynamics simulation studies of reactions involving the ethylene carbonate (EC) radical anion EC(-) using the reactive force field ReaxFF. Our studies reveal that the substantial barrier for transition from the closed (cyclic) form, denoted c-EC(-), of the radical anion to the linear (open) form, denoted o-EC(-), results in a relatively long lifetime of the c-EC(-) allowing this compound to react with other singly reduced alkyl carbonates. Using ReaxFF, we systematically investigate the fate of both c-EC(-) and o-EC(-) in the gas phase and EC solution. In the gas phase and EC solutions with a relatively low concentration of Li(+)/x-EC(-) (where x = o or c), radical termination reactions between radical pairs to form either dilithium butylene dicarbonate (CH(2)CH(2)OCO(2)Li)(2) (by reacting two Li(+)/o-EC(-)) or ester-carbonate compound (by reacting Li(+)/o-EC(-) with Li(+)/c-EC(-)) are observed. At higher concentrations of Li(+)/x-EC(-) in solution, we observe the formation of diradicals which subsequently lead to formation of longer alkyl carbonates oligomers through reaction with other radicals or, in some cases, formation of (CH(2)OCO(2)Li)(2) through elimination of C(2)H(4). We conclude that the local ionic concentration is important in determining the fate of x-EC(-) and that the reaction of c-EC(-) with o-EC(-) may compete with the formation of various alkyl carbonates from o-EC(-)/o-EC(-) reactions. read less NOT USED (low confidence) A. P. Garcia, D. Sen, and M. Buehler, “Nature’s Flexible and Tough Armor.” 2011. link Times cited: 1 NOT USED (low confidence) M. F. Russo and A. Duin, “Atomistic-scale simulations of chemical reactions: Bridging from quantum chemistry to engineering,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2011. link Times cited: 133 NOT USED (low confidence) O. Rahaman et al., “Development of a ReaxFF reactive force field for aqueous chloride and copper chloride.,” The journal of physical chemistry. A. 2010. link Times cited: 52 Abstract: Copper ions play crucial roles in many enzymatic and aqueous… read moreAbstract: Copper ions play crucial roles in many enzymatic and aqueous processes. A critical analysis of the fundamental properties of copper complexes is essential to understand their impact on a wide range of chemical interactions. However the study of copper complexes is complicated by the presence of strong polarization and charge transfer effects, multiple oxidation states, and quantum effects like Jahn-Teller distortions. These complications make the experimental observations difficult to interpret. In order to provide a computationally inexpensive yet reliable method for simulation of aqueous-phase copper chemistry, ReaxFF reactive force field parameters have been developed. The force field parameters have been trained against a large set of DFT-derived energies for condensed-phase copper-chloride clusters as well as chloride/water and copper-chloride/water clusters sampled from molecular dynamics (MD) simulations. The parameters were optimized by iteratively training them against configurations generated from ReaxFF MD simulations that are performed multiple times with improved sets of parameters. This cycle was repeated until the ReaxFF results were in accordance with the DFT-derived values. We have performed MD simulations on chloride/water and copper-chloride/water systems to validate the optimized force field. The structural properties of the chloride/water system are in accord with previous experimental and computational studies. The properties of copper-chloride/water agreed with the experimental observations including evidence of the Jahn-Teller distortion. The results of this study demonstrate the applicability of ReaxFF for the precise characterization of aqueous copper chloride. This force field provides a base for the design of a computationally inexpensive tool for the investigation of various properties and functions of metal ions in industrial, environmental, and biological environments. read less NOT USED (low confidence) C. R. Catlow, S. French, A. Sokol, A. Al-Sunaidi, and S. Woodley, “Zinc oxide: A case study in contemporary computational solid state chemistry,” Journal of Computational Chemistry. 2008. link Times cited: 101 Abstract: Computational techniques have been applied to study a broad … read moreAbstract: Computational techniques have been applied to study a broad range of chemical and physical properties of zinc oxide. Both interatomic‐potential and density functional theory methods are used to investigate structural, thermodynamic, surface, and defect properties. We survey the structures and energies of nano‐particulate zinc oxide. © 2008 Wiley Periodicals, Inc. J Comput Chem 2008 read less NOT USED (low confidence) S. Savka, A. Serednytski, and D. Popovych, “Modeling the adsorption processes and luminescence properties of the metal oxide ZnO nanoparticles,” Journal of Physical Studies. 2023. link Times cited: 0 Abstract: Modeling the adsorption processes and luminescence propertie… read moreAbstract: Modeling the adsorption processes and luminescence properties of Zinc Oxide (ZnO) can provide valuable insights into its applications. We used Molecular Dynamics (MD) method to investigate the adsorption processes on ZnO nanoclusters under di(cid:27)erent initial conditions. To ensure the nanoclusters were correctly structured, we applied Radial Distribution Functions (RDF) and Central Symmetry Parameter (CSP) methods. It was discovered that the number of defects in the samples had a major in(cid:29)uence on the simulated photoluminescence (PL) spectra, which were created using a bi-Gaussian function. To assess the amount of vacancies on the surfaces of the sample, we used the relative luminescence intensity of the secondary peak in the PL spectra. To analyze the simulated PL spectra, we utilized a Gaussian (cid:28)tting technique. The self-activated PL band peaking was divided by Gaussian deconvolution, which was utilized for a more in-depth analysis of the data. By researching the consequences of varying conditions on the PL spectra, we were able to obtain a better comprehension of the mechanisms behind adsorption processes on ZnO nanoclusters. Furthermore, this research enabled us to gain insight into the in(cid:29)uences that di(cid:27)erent conditions can have on the adsorption of oxygen atoms on the nanoclusters and helped us in creating new generation gas sensors based on ZnO nanopowders and its compounds. read less NOT USED (low confidence) R. Evarestov, “Binary Oxides of Transition Metals: ZnO, TiO$_2, ZrO_2, HfO_2$.” 2020. link Times cited: 0 NOT USED (low confidence) B. Cowen and M. El-Genk, “Bond-order reactive force fields for molecular dynamics simulations of crystalline silica,” Computational Materials Science. 2016. link Times cited: 15 NOT USED (high confidence) M. D. K. Jones, J. Dawson, S. Campbell, V. Barrioz, L. D. Whalley, and Y. Qu, “Modelling Interfaces in Thin-Film Photovoltaic Devices,” Frontiers in Chemistry. 2022. link Times cited: 2 Abstract: Developing effective device architectures for energy technol… read moreAbstract: Developing effective device architectures for energy technologies—such as solar cells, rechargeable batteries or fuel cells—does not only depend on the performance of a single material, but on the performance of multiple materials working together. A key part of this is understanding the behaviour at the interfaces between these materials. In the context of a solar cell, efficient charge transport across the interface is a pre-requisite for devices with high conversion efficiencies. There are several methods that can be used to simulate interfaces, each with an in-built set of approximations, limitations and length-scales. These methods range from those that consider only composition (e.g. data-driven approaches) to continuum device models (e.g. drift-diffusion models using the Poisson equation) and ab-initio atomistic models (developed using e.g. density functional theory). Here we present an introduction to interface models at various levels of theory, highlighting the capabilities and limitations of each. In addition, we discuss several of the various physical and chemical processes at a heterojunction interface, highlighting the complex nature of the problem and the challenges it presents for theory and simulation. read less NOT USED (high confidence) J. Goniakowski, S. Menon, G. Laurens, and J. Lam, “Nonclassical Nucleation of Zinc Oxide from a Physically Motivated Machine-Learning Approach,” The Journal of Physical Chemistry C. 2021. link Times cited: 3 Abstract: Observing non-classical nucleation pathways remains challeng… read moreAbstract: Observing non-classical nucleation pathways remains challenging in simulations of complex materials with technological interests. This is because it requires very accurate force fields that can capture the whole complexity of their underlying interatomic interactions and an advanced structural analysis. Here, we first report the construction of a machine-learning force field for zinc oxide interactions using the Physical LassoLars Interaction Potentials approach which allows us to be predictive even for untrained structures. Then, we carried out freezing simulations from a liquid and observed the crystal formation with atomistic precision. Our results, which are analyzed using a data-driven approach based on bond order parameters, demonstrate the presence of both prenucleation clusters and two-step nucleation scenarios thus retrieving seminal predictions of non-classical nucleation pathways made on much simpler models. read less NOT USED (high confidence) W. Brandão, A. L. Aguiar, and J. M. Sousa, “Atomistic Computational Modeling of Temperature Effects in Fracture Toughness and Degradation of Penta-graphene Monolayer.” 2021. link Times cited: 7 NOT USED (high confidence) E. Khare, N. Holten-Andersen, and M. Buehler, “Transition-metal coordinate bonds for bioinspired macromolecules with tunable mechanical properties,” Nature Reviews Materials. 2021. link Times cited: 96 NOT USED (high confidence) J. F. Dethan and V. Swamy, “Tensile properties of hydrogenated hybrid graphene–hexagonal boron nitride nanosheets: a reactive force field study,” Molecular Simulation. 2020. link Times cited: 4 Abstract: ABSTRACT Tensile properties of hydrogenated hybrid graphene–… read moreAbstract: ABSTRACT Tensile properties of hydrogenated hybrid graphene–hexagonal boron nitride (h-BN) nanosheets were investigated atomistically using molecular dynamics simulations in conjunction with a reactive force field (ReaxFF). ReaxFF was chosen to provide more reliable results compared to those of previous studies that used simpler atomistic potential models to predict mechanical properties. The hydrogenation site that provides the most significant reduction in Young’s modulus of the hybrid structures has been identified. The results show that fully hydrogenated hybrid nanosheet has the lowest Young’s modulus compared to partially hydrogenated or pristine configurations. It was also seen that hydrogenation reduces Young’s modulus of hybrid nanosheets regardless of the hydrogenation configuration. However, hydrogenated graphene was found to be stiffer than the equivalent hydrogenated BN structures on account of the stronger C–C bond compared to C–B, C–N and BN bonds. Significant differences in the deformation behaviour are observed for various hydrogenation regimes examined as also their temperature dependencies. Pristine hybrid nanosheet exhibits brittle failure whereas the hydrogenated hybrid nanosheets show ductile failures. The lowering of Young’s modulus due to hydrogenation of the hybrids becomes less significant with increasing temperature. It is expected that the present results provide useful knowledge for suggested applications of hydrogenated hybrids as nanodiodes and nanotransitors. read less NOT USED (high confidence) M. Izadi, A. Maghari, W. Zhang, and A. V. van Duin, “Reactive molecular dynamics simulation for isotope-exchange reactions in H/D systems: ReaxFFHD development.,” The Journal of chemical physics. 2020. link Times cited: 1 Abstract: To investigate the chemical isotope-exchange reactions withi… read moreAbstract: To investigate the chemical isotope-exchange reactions within a system composed of a mixture of hydrogen and deuterium (H/D) in the plasma media, the ReaxFFHD potential was parameterized against an appropriate quantum mechanics (QM)-based training set. These QM data involve structures and energies related to bond dissociation, angle distortion, and an exchange reaction of the tri-atomic molecular ions, H3 +, D3 +, H2D+, and D2H+, produced in the hydrogen plasma. Using the ReaxFFHD potential, a range of reactive molecular dynamics simulations were performed on different mixtures of H/D systems. Analysis of the reactions involved in the production of these tri-atomic molecular ions was carried out over 1 ns simulations. The results show that the ReaxFFHD potential can properly model isotope-exchange reactions of tri-atomic molecular ions and that it also has a perfect transferability to reactions taking place in these systems. In our simulations, we observed some intermediate molecules (H2, D2, and HD) that undergo secondary reactions to form the tri-atomic molecular ions as the most likely products in the hydrogen plasma. Moreover, there remains a preference for D in the produced molecular ions, which is related to the lower zero-point energy of the D-enriched species, showing the isotope effects at the heart of the ReaxFFHD potential. read less NOT USED (high confidence) M. Sengul, J. Guo, C. Randall, and A. C. T. van Duin, “Water-mediated surface diffusion mechanism enabling the Cold Sintering Process: A combined computational and experimental study.,” Angewandte Chemie. 2019. link Times cited: 44 Abstract: Cold Sintering Process (CSP) densifies ceramics at extremely… read moreAbstract: Cold Sintering Process (CSP) densifies ceramics at extremely low temperatures relative to conventional sintering processes. Several ceramics and composite systems have been successfully densified under cold sintering; in the case of zinc oxide grain growth kinetics, reduced activation energies are shown, and yet, the mechanism behind this growth is unknown. Herein, we investigate these mechanisms in more detail with experiments and ReaxFF molecular dynamics simulations. We investigated the recrystallization of zinc cations at various acidic conditions and found that its adsorption to surface can be a rate-limiting factor for cold sintering. Our studies show that surface hydroxylation in CSP does not inhibit crystallization; in contrast, by creating a surface complex, it creates an orders of magnitude acceleration in surface diffusion, and in turn, accelerate recrystallization. read less NOT USED (high confidence) L. Guo, Z. Zhou, L. Chen, S.-quan Shan, and Z. Wang, “Study of the pyrolysis of coals of different rank using the ReaxFF reactive force field,” Journal of Molecular Modeling. 2019. link Times cited: 13 NOT USED (high confidence) O. R. Gittus, G. von Rudorff, K. Rosso, and J. Blumberger, “Acidity Constants of the Hematite-Liquid Water Interface from Ab Initio Molecular Dynamics.,” The journal of physical chemistry letters. 2018. link Times cited: 18 Abstract: The interface between transition metal oxides (TMO) and liqu… read moreAbstract: The interface between transition metal oxides (TMO) and liquid water plays a crucial role in environmental chemistry, catalysis, and energy science. Yet, the mechanism and energetics of chemical transformations at solvated TMO surfaces is often unclear, largely because of the difficulty to characterize the active surface species experimentally. The hematite (α-Fe2O3)-liquid water interface is a case in point. Here we demonstrate that ab initio molecular dynamics is a viable tool for determining the protonation states of complex interfaces. The p Ka values of the oxygen-terminated (001) surface group of hematite, ≡OH, and half-layer terminated (012) surface groups, ≡2OH and ≡1OH2, are predicted to be (18.5 ± 0.3), (18.9 ± 0.6), and (10.3 ± 0.5) p Ka units, respectively. These are in good agreement with recent bond-valence theory based estimates, and suggest that the deprotonation of these surfaces require significantly more free energy input than previously thought. read less NOT USED (high confidence) A. L. Lloyd, Y.-hua Zhou, M. Yu, C. Scott, R. Smith, and S. Kenny, “Reaction pathways in atomistic models of thin film growth.,” The Journal of chemical physics. 2017. link Times cited: 8 Abstract: The atomistic processes that form the basis of thin film gro… read moreAbstract: The atomistic processes that form the basis of thin film growth often involve complex multi-atom movements of atoms or groups of atoms on or close to the surface of a substrate. These transitions and their pathways are often difficult to predict in advance. By using an adaptive kinetic Monte Carlo (AKMC) approach, many complex mechanisms can be identified so that the growth processes can be understood and ultimately controlled. Here the AKMC technique is briefly described along with some special adaptions that can speed up the simulations when, for example, the transition barriers are small. Examples are given of such complex processes that occur in different material systems especially for the growth of metals and metallic oxides. read less NOT USED (high confidence) A. Bandura, R. Evarestov, S. I. Lukyanov, S. Piskunov, and Y. Zhukovskii, “Simulation of Young’s moduli for hexagonal ZnO [0 0 0 1]-oriented nanowires: first principles and molecular mechanical calculations,” Materials Research Express. 2017. link Times cited: 10 Abstract: Morphologically reproducible wurtzite-structured zinc oxide … read moreAbstract: Morphologically reproducible wurtzite-structured zinc oxide nanowires (ZnO NWs) can be synthesized by different methods. Since ZnO NWs have been found to possess piezoelectricity, a comprehensive study of their mechanical properties, e.g. deformations caused by external compression or stretching, is one of the actual tasks of this paper. We have calculated wurtzite-structured [0 0 0 1]-oriented ZnO NWs whose diameters have been varied within 1–5 nm and 1–20 nm ranges when using either ab initio (hybrid DFT-LCAO) or force-field (molecular mechanical) methods, respectively (the minimum diameter dNW of experimentally synthesized NWs has been estimated on average to be ~20 nm). When using both chosen calculation approaches, the values of Young’s moduli determined for the mentioned ranges of NW diameters have been found to be qualitatively compatible (168–169 GPa for 5 nm NW thickness), whereas results of molecular mechanical simulations on YNW for 20 nm-thick NWs (160–162 GPa) have been qualitatively comparable with those experimentally measured along the [0 0 0 1] direction of NW loading. In all the cases, a gradual increase of the NW diameter has resulted in an asymptotic decrease of Young’s modulus consequently approaching that (Yb) of wurtzite-structured ZnO bulk along its [0 0 0 1] axis. The novelty of this study is that we combine the computation methods of quantum chemistry and molecular mechanics, while the majority of previous studies with the same aim have focused on the application of different classical molecular dynamical methods. read less NOT USED (high confidence) J. Wang, Y. Shen, F. Song, F. Ke, X. Liao, and C. Lu, “On the wurtzite to tetragonal phase transformation in ZnO nanowires,” Nanotechnology. 2017. link Times cited: 9 Abstract: There is a long standing contradiction on the tensile respon… read moreAbstract: There is a long standing contradiction on the tensile response of zinc oxide nanowires between theoretical prediction and experimental observations. Although it is proposed that there is a ductile behavior dominated by phase transformation, only an elastic deformation and brittle fracture was witnessed in experiments. Using molecular dynamics simulations, we clarified that, as the lateral dimension of zinc oxide nanowires increases to a critical value, an unambiguous ductile-to-brittle transition occurs. The critical value increases with decreasing the strain rate. Factors including planar defects and surface contamination induce brittle fracture prior to the initiation of phase transformation. These findings are consistent with previous atomistic standpoints and experimental results. read less NOT USED (high confidence) P. Li and K. Merz, “Metal Ion Modeling Using Classical Mechanics,” Chemical Reviews. 2017. link Times cited: 230 Abstract: Metal ions play significant roles in numerous fields includi… read moreAbstract: Metal ions play significant roles in numerous fields including chemistry, geochemistry, biochemistry, and materials science. With computational tools increasingly becoming important in chemical research, methods have emerged to effectively face the challenge of modeling metal ions in the gas, aqueous, and solid phases. Herein, we review both quantum and classical modeling strategies for metal ion-containing systems that have been developed over the past few decades. This Review focuses on classical metal ion modeling based on unpolarized models (including the nonbonded, bonded, cationic dummy atom, and combined models), polarizable models (e.g., the fluctuating charge, Drude oscillator, and the induced dipole models), the angular overlap model, and valence bond-based models. Quantum mechanical studies of metal ion-containing systems at the semiempirical, ab initio, and density functional levels of theory are reviewed as well with a particular focus on how these methods inform classical modeling efforts. Finally, conclusions and future prospects and directions are offered that will further enhance the classical modeling of metal ion-containing systems. read less NOT USED (high confidence) M. Ghahremanpour, P. V. Maaren, J. C. Ditz, R. Lindh, and D. Spoel, “Large-scale calculations of gas phase thermochemistry: Enthalpy of formation, standard entropy, and heat capacity,” Journal of Chemical Physics. 2016. link Times cited: 55 Abstract: Large scale quantum calculations for molar enthalpy of forma… read moreAbstract: Large scale quantum calculations for molar enthalpy of formation (Delta(f) H-0), standard entropy (S-0), and heat capacity (C-V) are presented. A large data set may help to evaluate quantum thermoc ... read less NOT USED (high confidence) A. Pathak, S. Nedea, A. V. van Duin, H. Zondag, C. Rindt, and D. Smeulders, “Reactive force field development for magnesium chloride hydrates and its application for seasonal heat storage.,” Physical chemistry chemical physics : PCCP. 2016. link Times cited: 21 Abstract: MgCl2 hydrates are considered as high-potential candidates f… read moreAbstract: MgCl2 hydrates are considered as high-potential candidates for seasonal heat storage materials. These materials have high storage capacity and fast dehydration kinetics. However, as a side reaction to dehydration, hydrolysis may occur. Hydrolysis is an irreversible reaction, which produces HCl gas thus affecting the durability of heat storage systems. In this study, we present the parameterization of a reactive force field (ReaxFF) for MgCl2 hydrates to study the dehydration and hydrolysis kinetics of MgCl2·H2O and MgCl2·2H2O. The ReaxFF parameters have been derived by training against quantum mechanics data obtained from Density Functional Theory (DFT) calculations consisting of bond dissociation curves, angle bending curves, reaction enthalpies, and equation of state. A single-parameter search algorithm in combination with a Metropolis Monte Carlo algorithm is successfully used for this ReaxFF parameterization. The newly developed force field is validated by examining the elastic properties of MgCl2 hydrates and the proton transfer reaction barrier, which is important for the hydrolysis reaction. The bulk moduli of MgCl2·H2O and MgCl2·2H2O obtained from ReaxFF are in close agreement with the bulk moduli obtained from DFT. A barrier of 20.24 kcal mol(-1) for the proton transfer in MgCl2·2H2O is obtained, which is in good agreement with the barrier (19.55 kcal mol(-1)) obtained from DFT. Molecular dynamics simulations using the newly developed ReaxFF on 2D-periodic slabs of MgCl2·H2O and MgCl2·2H2O show that the dehydration rate increases more rapidly with temperature in MgCl2·H2O than in MgCl2·2H2O, in the temperature range 300-500 K. The onset temperature of HCl formation, a crucial design parameter in seasonal heat storage systems, is observed at 340 K for MgCl2·H2O, which is in agreement with experiments. The HCl formation is not observed for MgCl2·2H2O. The diffusion coefficient of H2O through MgCl2·H2O is lower than through MgCl2·2H2O, and can become a rate-limiting step. The diffusion coefficient increases with temperature and follows the Arrhenius law both for MgCl2·H2O and MgCl2·2H2O. These results indicate the validity of the ReaxFF approach for studying MgCl2 hydrates and provide important atomistic-scale insight of reaction kinetics and H2O transport in these materials. read less NOT USED (high confidence) Y. Han, D. D. Jiang, J. Zhang, W. Li, Z. Gan, and J. Gu, “Development, applications and challenges of ReaxFF reactive force field in molecular simulations,” Frontiers of Chemical Science and Engineering. 2016. link Times cited: 87 NOT USED (high confidence) C. Tainter and G. Schatz, “Reactive Force Field Modeling of Zinc Oxide Nanoparticle Formation,” Journal of Physical Chemistry C. 2016. link Times cited: 11 Abstract: A ReaxFF reactive force field is developed and used for mole… read moreAbstract: A ReaxFF reactive force field is developed and used for molecular dynamics studies of reactions that occur when diethyl zinc is exposed to a graphene surface that has been functionalized with epoxide groups. From past experiments, it is known that these conditions lead to zinc oxide nanoparticle formation. Molecular dynamics simulations are used to provide atom-level detail into the nanoparticle formation process, including the mechanisms whereby oxygen is abstracted from the graphene surface, thus enabling condensation reactions in which multiple zinc-containing species form zinc oxide fragments and ultimately nanoparticles. Structural properties of the nanoparticles show nonstoichiometric zinc oxide structures with average coordination numbers of 3.6 around each zinc. Time-dependent density functional theory calculations show that the absorption spectra of these clusters are red-shifted by a few tenths of an electronvolt compared to that of a wurtzite crystal structure, representing transitions from oxy... read less NOT USED (high confidence) Z. Fan, “Molecular Simulations of Nanoscale Transformations in Ionic Semiconductor Nanocrystals.” 2016. link Times cited: 0 Abstract: The aim of the study described in this thesis is to obtain a… read moreAbstract: The aim of the study described in this thesis is to obtain a profound understanding of transformations in NCs at the atomic level, by performing molecular simulations for such transformations, and by comparing the simulation results with available experimental high resolution transmission electron microscopy (HRTEM) data to validate the simulations and to reveal underlying physical mechanisms. These transformations include structural and morphological transitions and cation exchange processes in ionic nanocrystals (II-VI and IV-VI semiconductors). The main simulation method used is classical Molecular Dynamics (MD) simulation. First principles density functional theory (DFT) calculations were used to develop empirical force fields that are able to accurately reproduce phase transitions. Using these newly developed force fields, large scaled classical MD simulations were carried out and linked to HRTEM experiments. The partially charged rigid ion model (PCRIM) was chosen for the force fields. This PCRIM approach has a simple functional form with a few number of parameters and has a clear physical meaning for ionic crystals. To simulate cation exchange in colloidal NC systems at the NC/solution interface, we used a combination of all-atom force fields and a coarse-grained model. In Chapter 2, an ab-initio based force field for ZnO is developed within the framework of the PCRIM approach. The values of the partial charges were determined by Bader charge analysis of DFT calculations on various ZnO phases. Beside Coulombic interactions, only short-ranged pairwise interatomic interactions were included. An initial guess of the parameters of the short-ranged pair potentials were first obtained by the lattice inversion method. The parameters were further adjusted by an ab-initio potential surface fitting procedure. The new ZnO force field has a very simple functional form is able to accurately reproduce several important physical properties of ZnO materials. These physical properties include the lattice parameters and phase stability of several ZnO polymorphs, as well as the elastic constants, bulk moduli, phonon dispersion, and melting points of wurtzite ZnO. The transition pressure of the wurtzite-to-rocksalt transition calculated with the force field equals 12.3 GPa, in agreement with experimental measurements and DFT calculations. A wurtzite-to-honeycomb phase transition is predicted at an uniaxial pressure of 8.8 GPa. We found a rational and effective way to derive force fields with simple functional forms for accurate simulations of phase transitions in ionic crystals. In Chapter 3, we developed a transferable force field for CdS-CdSe-PbSPbSe solid systems. The selection of the force field and the fitting procedure are similar to that of the ZnO force field in Chapter 2. The challenges when developing this force field were to maintain the transferability of this force field for four materials (CdS, CdSe, PbS, and PbSe) and to describe their mixed phases. This was solved by assuming that different cations/anions have the same values of the partial charges, and that shortranged interatomic interactions between two cations/anions are the same in different materials. For the mixed phases, DFT calculations of the mixed phases were included in both the training and validation sets. This work is the first step for further simulation studies of these II-VI and IV-VI semiconductor NCs and heteronanocrystals (HNCs). In Chapter 4, a thermally induced morphological and structural transition of CdSe NCs was investigated using MD simulations. In MD simulations, a CdSe nanosphere with the ZB structure transforms into a tetrapodlike morphology at 800 K. In a CdSe tetrapod, four WZ legs attach to the {111} facets of a tetrahedral ZB core. This transformation is achieved by a layer-by-layer slip of the ZB-{111} bilayer. Simulations show that the slips are mediated by the formation of Cd vacancies on the surface of the NCs to overcome the potentially large energy barriers associated with slip. The morphology of the annealed NCs is found to be temperature and size-dependent. An octapod-like morphology is found in NCs with a relatively large NC size and in a certain range of the heating temperature. Surprisingly, nanoscale transformations of CdSe NCs have been directly observed in HRTEM in situ heating experiments. Our findings provide a simple method to modify the morphology of ionic NCs and can potentially be used in the synthesis of branched NCs. The cation exchange process of PbSe/CdSe HNCs has been investigated by HRTEM in situ heating experiments in combination with MD simulations and DFT calculations in Chapter 5. In the HRTEM experiments, we bserved that Cd atoms in PbSe/CdSe nanodumbbells (CdSe rods with one or two PbSe tip(s)) are replaced by Pb atoms. The exchange rate depends on the heating temperature and the amount of Pb atoms present in the system. Sometimes, fully converted PbSe nanodumbells can be observed. MD simulations were performed to investigate the mechanism of this cation exchange process. It was found that the the CdSe domains near the PbSe/CdSe interfaces have significant structural disorder. These findings are in line with the experimental observation that the exchange process proceeds in a layer-by-layer fashion along the WZ- direction. We concluded that cation exchange in PbSe/CdSe HNCs is mediated by the local structural disorder which enables the formation of vacancies and accelerated the motion of cations. In Chapter 6, a coarse-grained psuedoligand model was introduced to simulate cation exchange in PbS colloidal NCs taking into account the cation-solvent interactions. Modelling colloidal NC systems including interactions with the solvent has long been a challenge due to the large system size and long time scales. Here, we incorporated the effects of ligands and solvents into negatively charged large spherical coarse-grained psuedoligands. MD simulations combining coarse-grained and all-atom models can successfully reproduce the cation exchange process in PbS colloidal NCs. Simulations show that the exchange rate and system equilibrium can be controlled by the temperature and by changing ligands. The exchange process is directly related to vacancy formation and the high mobility of Cd ions at the PbS/CdS interface. Our simulations also predict that high-pressure conditions will be beneficial for achieving fast exchange at elevated temperatures. Our coarse-grained model can be easily extended to other systems for the computational investigation of transformations in nanostructures. read less NOT USED (high confidence) M. Hellström, D. Spångberg, and K. Hermansson, “Treatment of delocalized electron transfer in periodic and embedded cluster DFT calculations: The case of Cu on ZnO (10
1¯ 0),” Journal of Computational Chemistry. 2015. link Times cited: 2 Abstract: We assess the consequences of the interface model—embedded‐c… read moreAbstract: We assess the consequences of the interface model—embedded‐cluster or periodic‐slab model—on the ability of DFT calculations to describe charge transfer (CT) in a particularly challenging case where periodic‐slab calculations indicate a delocalized charge‐transfer state. Our example is Cu atom adsorption on ZnO(10 1¯ 0), and in fact the periodic slab calculations indicate three types of CT depending on the adsorption site: full CT, partial CT, and no CT. Interestingly, when full CT occurs in the periodic calculations, the calculated Cu atom adsorption energy depends on the underlying ZnO substrate supercell size, since when the electron enters the ZnO it delocalizes over as many atoms as possible. In the embedded‐cluster calculations, the electron transferred to the ZnO delocalizes over the entire cluster region, and as a result the calculated Cu atom adsorption energy does not agree with the value obtained using a large periodic supercell, but instead to the adsorption energy obtained for a periodic supercell of roughly the same size as the embedded cluster. Different density functionals (of GGA and hybrid types) and basis sets (local atom‐centered and plane‐waves) were assessed, and we show that embedded clusters can be used to model Cu adsorption on ZnO(10 1¯ 0), as long as care is taken to account for the effects of CT. © 2015 Wiley Periodicals, Inc. read less NOT USED (high confidence) B. Narayanan, S. A. Deshmukh, S. Sankaranarayanan, and S. Ramanathan, “Strong correlations between structural order and passive state at water–copper oxide interfaces,” Electrochimica Acta. 2015. link Times cited: 9 NOT USED (high confidence) T. Milek and D. Zahn, “Molecular modeling of (100) and (000) zinc oxide surface growth from solution: islands, ridges and growth-controlling additives,” CrystEngComm. 2015. link Times cited: 12 Abstract: The mechanism of (100) and (000) zinc oxide surface growth f… read moreAbstract: The mechanism of (100) and (000) zinc oxide surface growth from ethanolic solution is investigated by molecular simulation. Growth steps are modelled at the maximum level of detail, i.e. by association of individual Zn2+ and OH− ions. Apart from structural relaxation, a mixed quantum/classical approach is used to explicitly study the proton-transfer reactions during crystal growth. Starting from idealized surfaces, we find that the (000) face evolves into rough landscapes composed of small islands separated by ~1 nm. On the other hand, the (100) growth front shows the formation of ridges encompassed by analogous 100 planes of the wurtzite structure. Contrary to idealized surface models, such rough surfaces obtained from explicit growth simulations enable us to identify considerable differences in both the binding site and energy for the association of growth-controlling additives. Using acetate and citrate ions as examples, we demonstrated the preferential association with peaks and kinks, respectively. read less NOT USED (high confidence) G. J. Soldano, F. Zanotto, and M. Mariscal, “Mechanical stability of zinc oxide nanowires under tensile loading: is wurtzite stable at the nanoscale?,” RSC Advances. 2015. link Times cited: 9 Abstract: The mechanical response of zinc oxide nanowires under uniaxi… read moreAbstract: The mechanical response of zinc oxide nanowires under uniaxial tensile loading is investigated by molecular dynamics and supported by density functional calculations. Previous theoretical works predict a stress-induced phase transition which has not been observed experimentally in zinc oxide nanowires up to date. Here, we report an explanation for such a discrepancy. Our simulations reveal brittle failure at room temperature without phase transformation, in agreement with experiments. Interestingly, we also find that if the temperature is raised to 600 K, the phase transition occurs. A detailed reaction mechanism is proposed. For the first time, the associated rate constant has been calculated. Based on these results we propose an experimental procedure to finally observe the predicted phase transformation. read less NOT USED (high confidence) É. Maras, N. Salles, R. Tétot, T. Ala‐Nissila, and H. Jónsson, “Improved tight-binding charge transfer model and calculations of energetics of a step on the rutile TiO2(110) surface,” Journal of Physical Chemistry C. 2015. link Times cited: 11 Abstract: A second-moment, tight-binding charge equilibration (SMTB-Q)… read moreAbstract: A second-moment, tight-binding charge equilibration (SMTB-Q) model for the atomic interactions in TiO2 is refined by comparison with results of density functional theory (DFT) calculations within t... read less NOT USED (high confidence) D. Fantauzzi, J. Bandlow, L. Sabo, J. Mueller, A. V. van Duin, and T. Jacob, “Development of a ReaxFF potential for Pt-O systems describing the energetics and dynamics of Pt-oxide formation.,” Physical chemistry chemical physics : PCCP. 2014. link Times cited: 53 Abstract: ReaxFF force field parameters describing Pt-Pt and Pt-O inte… read moreAbstract: ReaxFF force field parameters describing Pt-Pt and Pt-O interactions have been developed and tested. The Pt-Pt parameters are shown to accurately account for the chemical nature, atomic structures and other materials properties of bulk platinum phases, low and high-index platinum surfaces and nanoclusters. The Pt-O parameters reliably describe bulk platinum oxides, as well as oxygen adsorption and oxide formation on Pt(111) terraces and the {111} and {100} steps connecting them. Good agreement between the force field and both density functional theory (DFT) calculations and experimental observations is demonstrated in the relative surface free energies of high symmetry Pt-O surface phases as a function of the oxygen chemical potential, making ReaxFF an ideal tool for more detailed investigations of more complex Pt-O surface structures. Validation for its application to studies of the kinetics and dynamics of surface oxide formation in the context of either molecular dynamics (MD) or Monte Carlo simulations are provided in part by a two-part investigation of oxygen diffusion on Pt(111), in which nudged elastic band (NEB) calculations and MD simulations are used to characterize diffusion processes and to determine the relevant diffusion coefficients and barriers. Finally, the power of the ReaxFF reactive force field approach in addressing surface structures well beyond the reach of routine DFT calculations is exhibited in a brief proof-of-concept study of oxygen adsorbate displacement within ordered overlayers. read less NOT USED (high confidence) S. Wang et al., “New Ab Initio Based Pair Potential for Accurate Simulation of Phase Transitions in ZnO,” Journal of Physical Chemistry C. 2014. link Times cited: 42 Abstract: A set of interatomic pair potentials is developed for ZnO ba… read moreAbstract: A set of interatomic pair potentials is developed for ZnO based on the partially charged rigid ion model (PCRIM). The derivation of the potentials combines lattice inversion, empirical fitting, and ab initio energy surface fitting. We show that, despite the low number of parameters in this model (8), a wide range of physical properties is accurately reproduced using the new potential model. The calculated lattice parameters and elastic constants of ZnO in the wurtzite (WZ) phase, as well as the lattice parameters and stabilities of ZnO in other high-pressure and metastable phases, agree well with experiments and with density functional theory (DFT) calculations. The calculated transition pressure of the wurtzite-to-rocksalt (WZ-to-RS) transition is 12.3 GPa. A wurtzite-to-honeycomb (WZ-to-HC) phase transition induced by uniaxial pressure along the c-axis is simulated by means of molecular dynamics (MD) simulations. The WZ-to-HC transition takes place at an uniaxial pressure of 8.8 GPa while the reverse tr... read less NOT USED (high confidence) D. Machon et al., “Thermodynamics of nanoparticles: experimental protocol based on a comprehensive Ginzburg-Landau interpretation.,” Nano letters. 2014. link Times cited: 12 Abstract: The effects of surface and interface on the thermodynamics o… read moreAbstract: The effects of surface and interface on the thermodynamics of small particles require a deeper understanding. This step is crucial for the development of models that can be used for decision-making support to design nanomaterials with original properties. On the basis of experimental results for phase transitions in compressed ZnO nanoparticles, we show the limitations of classical thermodynamics approaches (Gibbs and Landau). We develop a new model based on the Ginzburg-Landau theory that requires the consideration of several terms, such as the interaction between nanoparticles, pressure gradients, defect density, and so on. This phenomenological approach sheds light on the discrepancies in the literature as it identifies several possible parameters that should be taken into account to properly describe the transformations. For the sake of clarity and standardization, we propose an experimental protocol that must be followed during high-pressure investigations of nanoparticles in order to obtain coherent, reliable data that can be used by the scientific community. read less NOT USED (high confidence) C. Becker, F. Tavazza, Z. Trautt, and R. B. D. Macedo, “Considerations for choosing and using force fields and interatomic potentials in materials science and engineering,” Current Opinion in Solid State & Materials Science. 2013. link Times cited: 196 NOT USED (high confidence) B. Narayanan, I. Reimanis, and C. Ciobanu, “Atomic-scale mechanism for pressure-induced amorphization of β-eucryptite,” Journal of Applied Physics. 2013. link Times cited: 14 Abstract: We present here a reactive force field based metadynamics st… read moreAbstract: We present here a reactive force field based metadynamics study of pressure-induced amorphization in β-eucryptite, a lithium aluminum silicate that exhibits negative thermal expansion, i.e., volumetric contraction upon heating. From our simulations, we found that β-eucryptite amorphizes under a moderate applied pressure of ∼3 GPa. A careful inspection of the amorphous phase showed that it contains AlO3, AlO4, AlO5, and SiO4 polyhedra, indicating clear short-range order. We have also identified the atomic-scale processes responsible for the amorphization of β-eucryptite. These processes are (a) tilting and distortion of tetrahedra centered at Al/Si, (b) change in atomic coordination around Al, and (c) disordering of Li atoms with the formation of Li-Li, Li-O, and Li-O-Li linkages. We discuss our results in the context of a possible general link between negative thermal expansion, radiation tolerance, and pressure-induced amorphization in flexible network structures. read less NOT USED (high confidence) T. Senftle, R. Meyer, M. Janik, and A. V. van Duin, “Development of a ReaxFF potential for Pd∕O and application to palladium oxide formation.,” The Journal of chemical physics. 2013. link Times cited: 71 Abstract: Oxide formation on palladium surfaces impacts the activity a… read moreAbstract: Oxide formation on palladium surfaces impacts the activity and selectivity of Pd-based catalysts, which are widely employed under oxygen rich operating conditions. To investigate oxidation processes over Pd catalysts at time and length scales inaccessible to quantum based computational methods, we have developed a Pd∕O interaction potential for the ReaxFF reactive force field. The parameters of the ReaxFF potential were fit against an extensive set of quantum data for both bulk and surface properties. Using the resulting potential, we conducted molecular dynamics simulations of oxide formation on Pd(111), Pd(110), and Pd(100) surfaces. The results demonstrate good agreement with previous experimental observations; oxygen diffusion from the surface to the subsurface occurs faster on the Pd(110) surface than on the Pd(111) and Pd(100) surfaces under comparable conditions at high temperatures and pressures. Additionally, we developed a ReaxFF-based hybrid grand canonical Monte Carlo∕molecular dynamics (GC-MC∕MD) approach to assess the thermodynamic stability of oxide formations. This method is used to derive a theoretical phase diagram for the oxidation of Pd935 clusters in temperatures ranging from 300 K to 1300 K and oxygen pressures ranging from 10(-14) atm to 1 atm. We observe good agreement between experiment and ReaxFF, which validates the Pd∕O interaction potential and demonstrates the feasibility of the hybrid GC-MC∕MD method for deriving theoretical phase diagrams. This GC-MC∕MD method is novel to ReaxFF, and is well suited to studies of supported-metal-oxide catalysts, where the extent of oxidation in metal clusters can significantly influence catalytic activity, selectivity, and stability. read less NOT USED (high confidence) W. Song and S.-jin Zhao, “Development of the ReaxFF reactive force field for aluminum–molybdenum alloy,” Journal of Materials Research. 2013. link Times cited: 10 Abstract: We have developed a reactive force field within the ReaxFF f… read moreAbstract: We have developed a reactive force field within the ReaxFF framework to accurately describe reactions involving aluminum–molybdenum alloy, which are part parameters of Al–O–Mo ternary system metastable intermolecular composites. The parameters are optimized from a training set, whose data come from density functional theory (DFT) calculations and experimental value, such as heat of formation, geometry data, and equation of states, which are reproduced well by ReaxFF. Body-centered cubic molybdenum’s surface energy, vacancy formation, and two transformational paths, Bain and trigonal paths are calculated to validate the ReaxFF ability describing the defects and deformations. Some structures’ elastic constant and phonon are calculated by DFT and ReaxFF to predict the structures’ mechanics and kinetic stability. All those results indicate that the fitted parameters can describe the energy difference of various structures under various circumstances and generally represent the diffusion property but cannot reproduce the elasticity and phonon spectra so well. read less NOT USED (high confidence) M. Zheng, X. Li, and L. Guo, “Algorithms of GPU-enabled reactive force field (ReaxFF) molecular dynamics.,” Journal of molecular graphics & modelling. 2013. link Times cited: 80 NOT USED (high confidence) C. Zou, A. Duin, and D. Sorescu, “Theoretical Investigation of Hydrogen Adsorption and Dissociation on Iron and Iron Carbide Surfaces Using the ReaxFF Reactive Force Field Method,” Topics in Catalysis. 2012. link Times cited: 49 NOT USED (high confidence) A. P. Garcia, N. Pugno, and M. Buehler, “Superductile, Wavy Silica Nanostructures Inspired by Diatom Algae,” Advanced Engineering Materials. 2011. link Times cited: 19 Abstract: Biology implements intriguing structural design principles t… read moreAbstract: Biology implements intriguing structural design principles that allow for attractive mechanical properties—such as high strength, toughness, and extensibility despite being made of weak and brittle constituents, as observed in biomineralized structures. For example, diatom algae contain nanoporous hierarchical silicified shells, called frustules, which provide mechanical protection from predators and virus penetration. These frustules generally have a morphology resembling honeycombs within honeycombs, meshes, or wavy shapes, and are surprisingly tough when compared to bulk silica, which is one of the most brittle materials known. However, the reason for its extreme extensibility has not been explained from a molecular level upwards. By carrying out a series of molecular dynamics simulations with the first principles‐based reactive force field ReaxFF, the mechanical response of the structures is elucidated and correlated with underlying deformation mechanisms. Specifically, it is shown that for wavy silica, unfolding mechanisms are achieved for increasing amplitude and allow for greater ductility of up to 270% strain. This mechanism is reminiscent to the uncoiling of hidden length from proteins that allows for enhanced energy dissipation capacity and, as a result, toughness. We report the development of an analytical continuum model that captures the results from atomistic simulations and can be used in multiscale models to bridge to larger scales. Our results demonstrate that tuning the geometric parameters of amplitude and width in wavy silica nanostructures are beneficial in improving the mechanical properties, including enhanced deformability, effectively overcoming the intrinsic shortcomings of the base material that features extreme brittleness. read less NOT USED (high confidence) J. Gale, P. Raiteri, and A. V. van Duin, “A reactive force field for aqueous-calcium carbonate systems.,” Physical chemistry chemical physics : PCCP. 2011. link Times cited: 83 Abstract: A new reactive force field has been derived that allows the … read moreAbstract: A new reactive force field has been derived that allows the modelling of speciation in the aqueous-calcium carbonate system. Using the ReaxFF methodology, which has now been implemented in the program GULP, calcium has been simulated as a fixed charge di-cation species in both crystalline phases, such as calcite and aragonite, as well as in the solution phase. Excluding calcium from the charge equilibration process appears to have no adverse effects for the simulation of species relevant to the aqueous environment. Based on this model, the speciation of carbonic acid, bicarbonate and carbonate have been examined in microsolvated conditions, as well as bulk water. When immersed in a droplet of 98 water molecules and two hydronium ions, the carbonate ion is rapidly converted to bicarbonate, and ultimately carbonic acid, which is formed as the metastable cis-trans isomer under kinetic control. Both first principles and ReaxFF calculations exhibit the same behaviour, but the longer timescale accessible to the latter allows the diffusion of the carbonic acid to the surface of the water to be observed, where it is more stable at the interface. Calcium carbonate is also examined as ion pairs in solution for both CaCO(3)(0)((aq)) and CaHCO(3)(+)((aq)), in addition to the (1014) surface in contact with water. read less NOT USED (high confidence) W.-J. Lee, J.-G. Chang, S. Ju, and C.-H. Lee, “Mechanism of local stress release in armchair single-wall zinc oxide nanotube under tensile loading,” Journal of Nanoparticle Research. 2011. link Times cited: 2 NOT USED (high confidence) B. Narayanan, A. V. van Duin, B. Kappes, I. Reimanis, and C. Ciobanu, “A reactive force field for lithium–aluminum silicates with applications to eucryptite phases,” Modelling and Simulation in Materials Science and Engineering. 2011. link Times cited: 50 Abstract: We have parameterized a reactive force field (ReaxFF) for li… read moreAbstract: We have parameterized a reactive force field (ReaxFF) for lithium–aluminum silicates using density functional theory (DFT) calculations of structural properties of a number of bulk phase oxides, silicates and aluminates, as well as of several representative clusters. The force field parameters optimized in this study were found to predict lattice parameters and heats of formation of selected condensed phases in excellent agreement with previous DFT calculations and with experiments. We have used the newly developed force field to study the eucryptite phases in terms of their thermodynamic stability and their elastic properties. We have found that (a) these ReaxFF parameters predict the correct order of stability of the three crystalline polymorphs of eucryptite, α, β and γ, and (b) that upon indentation, a new phase appears at applied pressures ⩾7 GPa. The high-pressure phase obtained upon indentation is amorphous, as illustrated by the radial distribution functions calculated for different pairs of elements. In terms of elastic properties analysis, we have determined the elements of the stiffness tensor for α- and β-eucryptite at the level of ReaxFF, and discussed the elastic anisotropy of these two polymorphs. Polycrystalline average properties of these eucryptite phases are also reported to serve as ReaxFF predictions of their elastic moduli (in the case of α-eucryptite), or as tests against values known from experiments or DFT calculations (β-eucrypite). The ReaxFF potential reported here can also describe well single-species systems (e.g. Li-metal, Al-metal and condensed phases of silicon), which makes it suitable for investigating structure and properties of suboxides, atomic-scale mechanisms responsible for phase transformations, as well as oxidation–reduction reactions. read less NOT USED (high confidence) J. Jelic, K. Reuter, and R. Meyer, “The Role of Surface Oxides in NOx Storage Reduction Catalysts,” ChemCatChem. 2010. link Times cited: 14 Abstract: The need for greater fuel economy in automotive vehicles has… read moreAbstract: The need for greater fuel economy in automotive vehicles has driven a surge in the development of diesel and lean-burn gasoline engines. Coupled with the increase in fuel efficiency, an increase in the air to fuel ratio also results in heightened production of NOx. [1] Several different strategies have been proposed for the handling of the higher NOx levels in lean-burn automotive emissions. One such strategy is the NOx storage reduction (NSR) system whereby excess NO is further oxidized to NO2 over the exhaust catalyst, which is typically platinum, and then NO2 is stored on a carrier material, such as BaO. [2] The catalyst system operates in this fashion for some minutes until the storage material has been fully converted. The system then cycles to a rich mode, during which a reductant is injected into the exhaust stream and reacts with the stored NOx, possibly at the interface between the carrier material and the metal particle, to produce the traditional exhaust products of N2, H2O, and CO2, all in a matter of seconds. Therefore, a requirement of a successful NSR catalyst is that the catalyst must be active for both NO oxidation and NOx reduction. In recent years, a debate has emerged as to the role of metal oxides in oxidation catalysis. [3–6] Although there have been some indications that oxide surfaces may be more active in simple oxidation reactions such as CO oxidation, [7] platinum catalysts have been preferred as the metal component in the NOx storage reduction system for their combination of high activity and ability to resist deactivation through oxidation. Ribeiro et al. have studied Pt NO-oxidation catalysts for NSR applications in considerable detail. They determined that large Pt particles are more active and that single crystal Pt(111) and Pt(100) outperform disperse Pt particles. [8, 9] One possible explanation for the higher activity of the single crystal surfaces is that oxygen is bound less strongly and that the catalysts do not deactivate due to oxide formation. Reports of platinum deactivation as a NO oxidation catalyst, however, concern catalysts that have been exposed to oxygen (and NO) for long periods. [9] As NSR systems operate under a cyclic operation read less NOT USED (high confidence) C. Catlow, S. Bromley, S. Hamad, M. Mora-Fonz, A. Sokol, and S. Woodley, “Modelling nano-clusters and nucleation.,” Physical chemistry chemical physics : PCCP. 2010. link Times cited: 160 Abstract: We review the growing role of computational techniques in mo… read moreAbstract: We review the growing role of computational techniques in modelling the structures and properties of nano-particulate oxides and sulphides. We describe the main methods employed, including those based on both electronic structure and interatomic potential approaches. Particular attention is paid to the techniques used in searching for global minima in the energy landscape defined by the nano-particle cluster. We summarise applications to the widely studied ZnO and ZnS systems, to silica nanochemistry and to group IV oxides including TiO(2). We also consider the special case of silica cluster chemistry in solution and its importance in understanding the hydrothermal synthesis of microporous materials. The work summarised, together with related experimental studies, demonstrates a rich and varied nano-cluster chemistry for these materials. read less NOT USED (high confidence) S. Savka, Y. I. Venhryn, A. Serednytski, and D. Popovych, “Molecular dynamics investigation of the formation processes of Zn–ZnO core-shell nanostructures,” Journal of Physical Studies. 2019. link Times cited: 3 NOT USED (high confidence) K. Sun, H.-Y. Su, and W. X. Li, “Stability of polar ZnO surfaces studied by pair potential method and local energy density method,” Theoretical Chemistry Accounts. 2013. link Times cited: 10 NOT USED (definite) A. Mishra et al., “Multiobjective genetic training and uncertainty quantification of reactive force fields,” npj Computational Materials. 2018. link Times cited: 25 NOT USED (definite) X. Zhang, Y. Wu, X. Chen, H. Wen, and S. Xiao, “Theoretical Study on Decomposition Mechanism of Insulating Epoxy Resin Cured by Anhydride,” Polymers. 2017. link Times cited: 33 Abstract: High temperatures caused by partial discharge results in the… read moreAbstract: High temperatures caused by partial discharge results in the decomposition of insulating epoxy resins in electrical equipment. In this paper, the ReaxFF force field is used to investigate the decomposition process of epoxy resins cured by anhydride and the formation mechanisms of small-molecule gases. Results show that the initiation reaction is the cleavage of an ester bond linked with an epoxy resin. Produced by the decomposition of ester groups, CO2 is the first and most abundant product. Meanwhile, CH2O can be generated through three main ways, although the process still depends on the decomposition of epoxy functional groups. H2O is produced by free radical collision and dehydration. The production of small-molecule gases has the following sequence: CO2, CH2O, CO, and H2O. The produced gases have the following order according to amount: CO2, CH2O, H2O, and CO. read less NOT USED (definite) T. Min, T. Yoon, C. Ling, S. Mahmud, T. Lim, and K. G. Saw, “Molecular dynamics simulations and photoluminescence measurements of annealed ZnO surfaces,” arXiv: Materials Science. 2016. link Times cited: 1 NOT USED (definite) B. Koo, N. Subramanian, and A. Chattopadhyay, “Molecular dynamics study of brittle fracture in epoxy-based thermoset polymer,” Composites Part B-engineering. 2016. link Times cited: 52 NOT USED (definite) A. L. Lloyd et al., “Development of a ReaxFF potential for Ag/Zn/O and application to Ag deposition on ZnO,” Surface Science. 2016. link Times cited: 29 NOT USED (definite) M. Hellström et al., “An SCC-DFTB Repulsive Potential for Various ZnO Polymorphs and the ZnO–Water System,” The Journal of Physical Chemistry. C, Nanomaterials and Interfaces. 2013. link Times cited: 38 Abstract: We have developed an efficient scheme for the generation of … read moreAbstract: We have developed an efficient scheme for the generation of accurate repulsive potentials for self-consistent charge density-functional-based tight-binding calculations, which involves energy-volume scans of bulk polymorphs with different coordination numbers. The scheme was used to generate an optimized parameter set for various ZnO polymorphs. The new potential was subsequently tested for ZnO bulk, surface, and nanowire systems as well as for water adsorption on the low-index wurtzite (101̅0) and (112̅0) surfaces. By comparison to results obtained at the density functional level of theory, we show that the newly generated repulsive potential is highly transferable and capable of capturing most of the relevant chemistry of ZnO and the ZnO/water interface. read less NOT USED (definite) W.-J. Lee, J.-G. Chang, S. Ju, M. Weng, and C.-H. Lee, “Structure-dependent mechanical properties of ultrathin zinc oxide nanowires,” Nanoscale Research Letters. 2011. link Times cited: 23 |