Citations
This panel presents information regarding the papers that have cited the interatomic potential (IP) whose page you are on.
The OpenKIM machine learning based Deep Citation framework is used to determine whether the citing article actually used the IP in computations (denoted by "USED") or only provides it as a background citation (denoted by "NOT USED"). For more details on Deep Citation and how to work with this panel, click the documentation link at the top of the panel.
The word cloud to the right is generated from the abstracts of IP principle source(s) (given below in "How to Cite") and the citing articles that were determined to have used the IP in order to provide users with a quick sense of the types of physical phenomena to which this IP is applied.
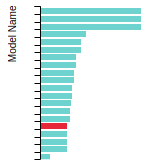
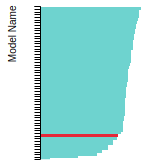
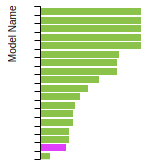
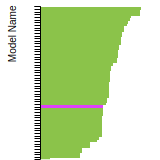
The bar chart shows the number of articles that cited the IP per year. Each bar is divided into green (articles that USED the IP) and blue (articles that did NOT USE the IP).
Users are encouraged to correct Deep Citation errors in determination by clicking the speech icon next to a citing article and providing updated information. This will be integrated into the next Deep Citation learning cycle, which occurs on a regular basis.
OpenKIM acknowledges the support of the Allen Institute for AI through the Semantic Scholar project for providing citation information and full text of articles when available, which are used to train the Deep Citation ML algorithm.
|
This panel provides information on past usage of this interatomic potential (IP) powered by the OpenKIM Deep Citation framework. The word cloud indicates typical applications of the potential. The bar chart shows citations per year of this IP (bars are divided into articles that used the IP (green) and those that did not (blue)). The complete list of articles that cited this IP is provided below along with the Deep Citation determination on usage. See the Deep Citation documentation for more information.

348 Citations (245 used)
Help us to determine which of the papers that cite this potential actually used it to perform calculations. If you know, click the .
USED (high confidence) I. Bejenari, A. Burenkov, P. Pichler, I. Deretzis, A. Sciuto, and A. L. Magna, “Molecular dynamics simulations supporting the development of a continuum model of heat transport in nanowires,” 2021 27th International Workshop on Thermal Investigations of ICs and Systems (THERMINIC). 2021. link Times cited: 0 Abstract: We establish a suitable methodology for Molecular Dynamics (… read moreAbstract: We establish a suitable methodology for Molecular Dynamics (MD) simulations to provide reliable data for the development of continuum model extensions of Fourier’s law, which reproduce effects arising from phonon confinement and interface scattering. This continuum approach for thermal transport is required for TCAD tools dedicated to nanoscaled electron device simulations. read less USED (high confidence) L. Zhang et al., “Toward Optimal Heat Transfer of 2D-3D Heterostructures via van der Waals Binding Effects.,” ACS applied materials & interfaces. 2021. link Times cited: 12 Abstract: Two-dimensional (2D) materials and their heterogeneous integ… read moreAbstract: Two-dimensional (2D) materials and their heterogeneous integration have enabled promising electronic and photonic applications. However, significant thermal challenges arise due to numerous van der Waals (vdW) interfaces limiting the dissipation of heat generated in the device. In this work, we investigate the vdW binding effect on heat transport through a MoS2-amorphous silica heterostructure. We show using atomistic simulations that the cross-plane thermal conductance starts to saturate with the increase of vdW binding energy, which is attributed to substrate-induced localized phonons. With these atomistic insights, we perform device-level heat transfer optimizations. Accordingly, we identify a regime, characterized by the coupling of in-plane and cross-plane heat transport mediated by vdW binding energy, where maximal heat dissipation in the device is achieved. These results elucidate fundamental heat transport through the vdW heterostructure and provide a pathway toward optimizing thermal management in 2D nanoscale devices. read less USED (high confidence) M. R. Vazirisereshk, S. A. Sumaiya, R. Chen, M. Baykara, and A. Martini, “Time-Dependent Electrical Contact Resistance at the Nanoscale,” Tribology Letters. 2021. link Times cited: 4 USED (high confidence) K. Sun, J. Chen, B.-jie Wu, L. Wang, and L. Fang, “Size-Dependent Mechanical Properties of Amorphous SiO2 Nanowires: A Molecular Dynamics Study,” Materials. 2020. link Times cited: 1 Abstract: Uniaxial tension tests were performed for amorphous SiO2 nan… read moreAbstract: Uniaxial tension tests were performed for amorphous SiO2 nanowires using molecular dynamics simulation to probe the size effect on the mechanical properties and plastic deformation by varying the length of nanowires. The simulation results showed that the Young’s modulus of SiO2 nanowires increased with the decrease of nanowires length due to its higher surface stress. The corresponding deformation of SiO2 nanowires during tension exhibited two periods: atomic arrangement at small strain and plastic deformation at large strain. During the atomic arrangement period, the percentage variations of atom number of 2-coordinated silicon and 3-coordinated silicon (PCN2 and PCN3) decreased, while the percentage variations of atom number of 4-coordinated silicon, 5-coordinated silicon (PCN4 and PCN5) and the Si–O bond number (PCB) rose slightly with increasing strain, as the strain was less than 22%. The situation reversed at the plastic deformation period, owing to the numerous breakage of Si–O bonds as the strain grew beyond 22%. The size effect of nanowires radius was considered, finding that the Young’s modulus and fracture stress were higher for the larger nanowire because of fewer dangling bonds and coordinate defeats in the surface area. The elastic deformation occurred at a small strain for the larger nanowire, followed by the massive plastic deformation during tension. A brittle mechanism covers the fracture characteristics, irrespective of the nanowire size. read less USED (high confidence) H. G. Ozcelik, E. Satiroglu, and M. Barisik, “Size dependent influence of contact line pinning on wetting of nano-textured/patterned silica surfaces.,” Nanoscale. 2020. link Times cited: 8 Abstract: Wetting behavior on a heterogeneous surface undergoes contac… read moreAbstract: Wetting behavior on a heterogeneous surface undergoes contact angle hysteresis as the droplet stabilized at a metastable state with a contact angle significantly different from its equilibrium value due to contact line pinning. However, there is a lack of consensus on how to calculate the influence of pinning forces. In general, the pinning effect can be characterized as (i) microscopic behavior when a droplet is pinned and the contact angle increases/decreases as the droplet volume increases/decreases and (ii) macroscopic behavior as the pinning effects decrease and ultimately, disappear with the increase of the droplet size. The current work studied both behaviors using molecular dynamics (MD) simulation with more than 300 different size water droplets on silica surfaces with three different patterns across two different wetting conditions. Results showed that the contact angle increases linearly with increasing droplet volume through the microscopic behavior, while the droplet is pinned on top of a certain number of patterns. When we normalized the droplet size with the corresponding pattern size, we observed a "wetting similarity" that linear microscopic contact angle variations over different size heterogeneities continuously line up. This shows that the pinning force remains constant and the resulting pinning effects are scalable by the size ratio between the droplet and pattern, independent of the size-scale. The slope of these microscopic linear variations decreases with an increase in the droplet size as observed through the macroscopic behavior. We further found a universal behavior in the variation of the corresponding pinning forces, independent of the wetting condition. In macroscopic behavior, pinning effects become negligible and the contact angle reaches the equilibrium value of the corresponding surface when the diameter of the free-standing droplet is approximately equal to 24 times the size of the surface structure. We found that the pinning effect is scalable with the droplet volume, not the size of the droplet base. read less USED (high confidence) D. Martí, J. Ainsley, Ó. Ahumada, C. Alemán, and J. Torras, “Tethering of the IgG1 Antibody to Amorphous Silica for Immunosensor Development: A Molecular Dynamics Study.,” Langmuir : the ACS journal of surfaces and colloids. 2020. link Times cited: 4 Abstract: A key factor for improving the sensitivity and performance o… read moreAbstract: A key factor for improving the sensitivity and performance of immunosensors based on mechanical-plasmonic methods is the orientation of the antibody proteins immobilized on the inorganic surface. Although experimental techniques fail to determine surface phenomena at the molecular level, modern simulations open the possibility for improving our understanding of protein-surface interactions. In this work, replica exchange molecular dynamics (REMD) simulations have been used to model the IgG1 protein tethered onto the amorphous silica surface by considering a united-atom model and a relatively large system (2500 nm2 surface). Additional molecular dynamics (MD) simulations have been conducted to derive an atomistic model for the amorphous silica surface using the cristobalite crystal structure as a starting point and to examine the structure of the free IgG1 antibody in the solution for comparison when immobilized. Analyses of the trajectories obtained for the tethered IgG1, which was sampled considering 32 different temperatures, have been used to define the geometry of the protein with respect to the inorganic surface. The tilt angle of the protein with respect to the surface plane increases with temperature, the most populated values being 24, 66, and 87° at the lowest (250 K), room (298 K), and the highest (380 K) temperatures. This variation indicates that the importance of protein-surface interactions decreases with increasing temperature. The influence of the surface on the structure of the antibody is very significant in the constant region, which is directly involved in the tethering process, while it is relatively unimportant for the antigen-binding fragments, which are farthest from the surface. These results are expected to contribute to the development of improved mechanical-plasmonic sensor microarrays in the near future. read less USED (high confidence) H. Amekura et al., “On the mechanism of the shape elongation of embedded nanoparticles,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2020. link Times cited: 9 USED (high confidence) A. Gabourie, S. Suryavanshi, A. Farimani, and E. Pop, “Reduced thermal conductivity of supported and encased monolayer and bilayer MoS2,” 2D Materials. 2020. link Times cited: 28 Abstract: Electrical and thermal properties of atomically thin two-dim… read moreAbstract: Electrical and thermal properties of atomically thin two-dimensional (2D) materials are affected by their environment, e.g. through remote phonon scattering or dielectric screening. However, while it is known that mobility and thermal conductivity (TC) of graphene are reduced on a substrate, these effects are much less explored in 2D semiconductors such as MoS2. Here, we use molecular dynamics to understand TC changes in monolayer (1L) and bilayer (2L) MoS2 by comparing suspended, supported, and encased structures. The TC of monolayer MoS2 is reduced from ∼117 W m−1 K−1 when suspended, to ∼31 W m−1 K−1 when supported by SiO2, at 300 K. Encasing 1L MoS2 in SiO2 further reduces its TC down to ∼22 W m−1 K−1. In contrast, the TC of 2L MoS2 is not as drastically reduced, being >50% higher than 1L both when supported and encased. These effects are due to phonon scattering with remote vibrational modes of the substrate, which are partly screened in 2L MoS2. We also examine the TC of 1L MoS2 across a wide range of temperatures (300 K to 700 K) and defect densities (up to 5 × 1013 cm−2), finding that the substrate reduces the dependence of TC on these factors. Taken together, these are important findings for all applications which will use 2D semiconductors supported or encased by insulators, instead of freely suspended. read less USED (high confidence) G. Guttormsen, A. Fletcher, and M. Oppenheim, “Atomic‐Scale Simulations of Meteor Ablation,” Journal of Geophysical Research: Space Physics. 2020. link Times cited: 2 Abstract: Meteoroids smaller than a microgram constantly bombard the E… read moreAbstract: Meteoroids smaller than a microgram constantly bombard the Earth, depositing material in the mesosphere and lower thermosphere. Meteoroid ablation, the explosive evaporation of meteoroids due to erosive impacts of atmospheric particles, consists of sputtering and thermal ablation. This paper presents the first atomic‐scale modeling of sputtering, the initial stage of ablation where hypersonic collisions between the meteoroid and atmospheric particles cause the direct ejection of atoms from the meteoroid surface. Because meteoroids gain thermal energy from these particle impacts, these interactions are important for thermal ablation as well. In this study, a molecular dynamics simulator calculates the energy distribution of the sputtered particles as a function of the species, velocity, and angle of the incoming atmospheric particles. The sputtering yield generally agrees with semi‐empirical equations at normal incidence but disagrees with the generally accepted angular dependence. Λ, the fraction of energy from a single atmospheric particle impact incorporated into the meteoroid, was found to be less than 1 and dependent on the velocity, angle, atmospheric species, and meteoroid material. Applying this new Λ to an ablation model results in a slower meteoroid temperature increase and mass loss rate as a function of altitude. This alteration results in changes in the expected electron line densities and visual magnitudes of meteoroids. Notably, this analysis leads to the prediction that meteoroids will generally ablate 1–4 km lower than previously predicted. This affects analysis of radar and visual measurements, as well as determination of meteoroid mass. read less USED (high confidence) T. Kodama et al., “Modulation of Interfacial Thermal Transport between Fumed Silica Nanoparticles by Surface Chemical Functionalization for Advanced Thermal Insulation.,” ACS applied materials & interfaces. 2020. link Times cited: 8 Abstract: Since solid-state heat transport in a highly porous nanocomp… read moreAbstract: Since solid-state heat transport in a highly porous nanocomposite strongly depends on the thermal boundary conductance (TBC) between constituent nanomaterials, further suppression of the TBC is important for improving performance of thermal insulators. Here, targeting a nanocomposite fabricated by stamping fumed silica nanoparticles, we perform a wide variety of surface functionalizations on fumed silica nanoparticles by a silane coupling method and investigate the impact on the thermal conductivity (Km). The Km of the silica nanocomposite is approximately 20 and 9 mW/m/K under atmospheric and vacuum conditions at the material density of 0.2 g/cm3 without surface functionalization, respectively, and the experimental results indicate that the Km can be modulated depending on the chemical structure of molecules. The surface modification with a linear alkyl chain of optimal length significantly suppresses Km by approximately 30%, and the suppression can be further enhanced to approximately 50% with an infrared opacifier. The magnitude of suppression was found to sensitively depend on the length of the terminal chain. The magnitude is also related to the number of reactive silanol groups in the chemical structure, where the surface modification with fluorocarbon gives the largest suppression. The surface hydrophobization merits thermal insulation through significant suppression of the TBC, presumably by reducing the water molecules that otherwise would serve as heat conduction channels at the interface. On the other hand, when the chain length is long, the suppression is counteracted by the enhanced phonon transmission through the silane coupling molecules that grow with the chain length. This is supported by the analytical model and present simulation results, leading to prediction of the optimal chemical structure for better thermal insulation. read less USED (high confidence) Y. Liao and J. Shiomi, “Akhiezer mechanism dominates relaxation of propagons in amorphous material at room temperature,” Journal of Applied Physics. 2020. link Times cited: 4 Abstract: Propagons play an important role in tuning the thermal condu… read moreAbstract: Propagons play an important role in tuning the thermal conductivity of nanostructured amorphous materials. Although advances have been made to quantitatively evaluate the relaxation time of propagons with molecular dynamics, the underlying relaxation mechanism remains unexplored. Here, we investigate the relaxation process of propagons in amorphous silicon, amorphous silica, and amorphous silicon nitride at room temperature in terms of Akhiezer model, the parameters of which were evaluated by performing lattice dynamics and molecular dynamics analysis. The results show that the Akhiezer model can well reproduce experimental results obtained by various kinds of measurement methods, indicating that Akhiezer mechanism dominates the relaxation process of propagons at room temperature. Moreover, we show that the appropriate sound speed of propagons is around 80% of the Debye sound speed and comparable to that of the sound speed of the transversal modes. We also reveal that the contribution of diffusons to the total thermal conductivity of these amorphous is similar, which is around 1 W/m K, while the contribution of propagons varies significantly depending on the materials, which is 30% in amorphous silicon and silica but can be as high as 70% in amorphous silicon nitride. read less USED (high confidence) P. Desmarchelier, K. Termentzidis, and A. Tanguy, “Vibrational density of states of free and embedded semiconducting GaN nanoparticles,” Semiconductor Science and Technology. 2020. link Times cited: 2 Abstract: The impact of the size of free and embedded GaN nanoparticle… read moreAbstract: The impact of the size of free and embedded GaN nanoparticles on vibrational properties has been studied using three different numerical methods. The thermal conductivity of free nanoparticles was also estimated with equilibrium molecular dynamics. Important discrepancies between the vibrational density of states of small nanoparticles compared to the bulk are observed, such as the presence of modes in the bandgap related to the surface modes, the optical peaks decrease, and the redshift of the transverse acoustic peak. When these nanoparticles are embedded in a SiO2 matrix, the peaks in the bandgap disappear and the transverse acoustic modes are shifted back to the bulk frequencies. These differences between the free and the embedded nanoparticles tend to disappear for nanoparticles with diameters larger than 5 nm. Finally, the thermal conductivity for free nanoparticles is computed, showing a non-linear augmentation upon the increase of the size of nanoparticles. The latter results could be useful in effective medium models used to estimate the thermal conductivity of nanocomposites. read less USED (high confidence) H. Ozcelik, Y. Sozen, H. Sahin, and M. Barisik, “Parametrizing nonbonded interactions between silica and water from first principles,” Applied Surface Science. 2020. link Times cited: 6 USED (high confidence) J. Zhang et al., “Effects of interlayer interactions on the nanoindentation response of freely suspended multilayer gallium telluride,” Nanotechnology. 2019. link Times cited: 11 Abstract: Freestanding indentation is a widely used method to characte… read moreAbstract: Freestanding indentation is a widely used method to characterise the elastic properties of two-dimensional (2D) materials. However, many controversies and confusion remain in this field due to the lack of appropriate theoretical models in describing the indentation responses of 2D materials. Taking the multilayer gallium telluride (GaTe) as an example, in this paper we conduct a series of experiments and simulations to achieve a comprehensive understanding of its freestanding indentation behaviours. Specifically, the freestanding indentation experiments show that the elastic properties of the present multilayer GaTe with a relatively large thickness can only be extracted from the bending stage in the indentation process rather than the stretching stage widely utilised in the previous studies on thin 2D materials, since the stretching stage of thick 2D materials is inevitably accompanied with severe plastic deformations. In combination with existing continuum mechanical models and finite element simulations, an extremely small Young’s modulus of multilayer GaTe is obtained from the nanoindentation experiments, which is two orders of magnitude smaller than the value obtained from first principles calculations. Our molecular dynamics (MD) simulations reveal that this small Young’s modulus can be attributed to the significant elastic softening in the multilayer GaTe with increasing thickness and decreasing length. It is further revealed in MD simulations that this size-induced elastic softening originates from the synergistic effects of interlayer compression and interlayer shearing in the multilayer GaTe, both of which, however, are ignored in the existing indentation models. To consider these effects of interlayer interactions in the theoretical modelling of the freestanding indentation of multilayer GaTe, we propose here novel multiple-beam and multiple-plate models, which are found to agree well with MD results without any additional parameters fitting and thus can be treated as more precise theoretical models in characterising the freestanding indentation behaviours of 2D materials. read less USED (high confidence) N. A. Mehta and D. Levin, “Multiscale modeling of damaged surface topology in a hypersonic boundary.,” The Journal of chemical physics. 2019. link Times cited: 7 Abstract: In this work, we used molecular dynamics (MD) to perform tra… read moreAbstract: In this work, we used molecular dynamics (MD) to perform trajectory simulations of ice-like argon and amorphous silica aggregates on atomically smooth highly ordered pyrolytic graphite (HOPG) and a comparatively rougher quartz surface. It was found that at all incidence velocities, the quartz surface was stickier than the HOPG surface. The sticking probabilities and elastic moduli obtained from MD were then used to model surface evolution at a micron length scale using kinetic Monte Carlo (kMC) simulations. Rules were derived to control the number of sites available for the process execution in kMC to accurately model erosion of HOPG by atomic oxygen (AO) attack and ice-nucleation on surfaces. It was observed that the effect of defects was to increase the material erosion rate, while that of aggregate nucleation was to lower it. Similarly, simulations were performed to study the effects of AO attack and N2 adsorption-desorption on surface evolution and it was found that N2 adsorption-desorption limits the surface available for erosion by AO attack. read less USED (high confidence) Z. Chen, Y. Cao, W. Tian, and Y. Wang, “Surface roughness analysis of Cu films deposited on Si substrates: A molecular dynamic analysis,” Journal of Applied Physics. 2019. link Times cited: 4 Abstract: Cu is a promising material to replace Al and Au in integrate… read moreAbstract: Cu is a promising material to replace Al and Au in integrated circuits and microscale devices because of its low electrical resistivity, high electromigration resistance, and low cost. However, surface roughness affects the contact resistance of these devices, especially when the device is on a microscale or nanoscale. This paper focuses on surface roughness analysis of Cu films deposited on Si substrates by molecular dynamic simulation based on the mechanism of physical vapor deposition. The effects of film thickness, deposition temperature, deposition interval, and reflow temperature on the surface roughness of Cu films are studied in detail. The simulation results show that the surface roughness can be improved by appropriate adjustments of these parameters. They also provide a foundation for further work on the deposition of Cu films on Si substrates.Cu is a promising material to replace Al and Au in integrated circuits and microscale devices because of its low electrical resistivity, high electromigration resistance, and low cost. However, surface roughness affects the contact resistance of these devices, especially when the device is on a microscale or nanoscale. This paper focuses on surface roughness analysis of Cu films deposited on Si substrates by molecular dynamic simulation based on the mechanism of physical vapor deposition. The effects of film thickness, deposition temperature, deposition interval, and reflow temperature on the surface roughness of Cu films are studied in detail. The simulation results show that the surface roughness can be improved by appropriate adjustments of these parameters. They also provide a foundation for further work on the deposition of Cu films on Si substrates. read less USED (high confidence) J. Zhang et al., “Effects of grain boundary on wear of graphene at the nanoscale: A molecular dynamics study,” Carbon. 2019. link Times cited: 42 USED (high confidence) A. Rajabpour, S. Bazrafshan, and S. Volz, “Carbon-nitride 2D nanostructures: thermal conductivity and interfacial thermal conductance with the silica substrate.,” Physical chemistry chemical physics : PCCP. 2019. link Times cited: 36 Abstract: The rate of heat dissipation from a 2D nanostructure strongl… read moreAbstract: The rate of heat dissipation from a 2D nanostructure strongly depends on the interfacial thermal conductance with its substrate. In this paper, the interfacial thermal conductance of carbon-nitride 2D nanostructures (C3N, C2N, C3N4's) with silica substrates was investigated using transient molecular dynamics simulations. It was found that a 2D nanostructure with higher thermal conductivity, has a lower value of interfacial thermal conductance with the silica substrate. The thermal conductivity of suspended carbon-nitride 2D nanostructures was also calculated using the Green-Kubo formalism and compared with that of graphene as a reference structure. It was found that the thermal conductivities of C3N, C2N, C3N4 (s-triazine) and C3N4 (tri-triazine) are respectively 62%, 4%, 4% and 2% that of graphene; while their interfacial thermal conductances with silica are 113%, 171%, 212% and 188% that of graphene. These different behaviors of the thermal conductivity and the interfacial thermal conductance with the substrate may be important in the thermal management of carbon-nitride 2D nanostructures in nanoelectronics. read less USED (high confidence) T. Jiang, Z. Wang, X. Ruan, and Y. Zhu, “Equi-biaxial compressive strain in graphene: Grüneisen parameter and buckling ridges,” 2D Materials. 2018. link Times cited: 19 Abstract: Strain and defects in graphene have critical impact on morph… read moreAbstract: Strain and defects in graphene have critical impact on morphology and properties of graphene. Here we report equi-biaxial compressive strain in monolayer graphene on SiO2 and Si3N4 substrates induced by thermal cycling in vacuum. The equi-biaxial strain is attributed to the mismatch in coefficient of thermal expansion between graphene and the substrate and sliding of graphene on the substrate. The sliding occurs during heating at the temperatures of 390 and 360 K for graphene on SiO2 and Si3N4 substrates, respectively. The biaxial Grüneisen parameter is determined to be 1.95 and 3.15 for G and 2D Raman bands of graphene, respectively. As the heating temperature exceeds a threshold temperature (1040 K for graphene/SiO2 and 640 K for graphene/Si3N4), buckling ridges are observed in graphene after the thermal cycle, from which the biaxial buckling strain of graphene on SiO2 and Si3N4 substrates are obtained as 0.21% and 0.22%, respectively. Importantly, the induced buckling ridges in graphene exhibit a pattern representing the symmetry of graphene crystal structure, which indicates that graphene relieves the compressive stress mainly along its lattice symmetry directions. These thermally induced graphene ridges are also found reminiscent of those in the synthesized graphene, suggesting the same origin of formation of the buckling ridges under biaxial compression. read less USED (high confidence) N. Liao, H. Zhou, B. Zheng, and W. Xue, “Silicon Oxycarbide-Derived Carbon as Potential NO2 Gas Sensor: A First Principles’ Study,” IEEE Electron Device Letters. 2018. link Times cited: 34 Abstract: Silicon oxycarbide (SiCO)-derived porous carbon is a novel c… read moreAbstract: Silicon oxycarbide (SiCO)-derived porous carbon is a novel class of nano-porous material with unique properties including highly sensitive gas detection. In this letter, SiCO-derived porous carbon (porous SiCO) structures are successfully reproduced by simulating the etching process in experiments. Then, the gas sensing performance of SiCO-derived carbon with different porous morphologies is investigated. The calculated adsorption energy, Mulliken charge transfer, bandgap, and adsorption distance indicate that SiCO-derived porous carbon exhibits a higher sensitivity toward NO2 gas than CO, 2, and acetone in accordance with experimental conclusions. Moreover, the porous SiCO with the largest SSA and PV shows the most excellent NO2 sensing performance. The adsorption of NO2 leads to the appearance of a new strong peak at the edge of the conduction band, resulting in obvious changes of the conductivity of the systems, which is necessary for NO2 detection. read less USED (high confidence) H. Amekura et al., “Vaporlike phase of amorphous
SiO2
is not a prerequisite for the core/shell ion tracks or ion shaping,” Physical Review Materials. 2018. link Times cited: 9 Abstract: The SHI irradiations were performed under the CommonUse Faci… read moreAbstract: The SHI irradiations were performed under the CommonUse Facility Program of JAEA. H.A. was supported by JSPSKAKENHI Grant No. 18K04898. I.S., V.J., F.D., and K.N.
gratefully acknowledge financial support from the Academy
of Finland MESIOS and NANOIS projects, and CPU capacity
grants from the IT Centre for Science CSC in Espoo, Finland. Part of this work was performed at the SAXS/WAXS
beamline at the Australian Synchrotron, part of ANSTO. P.K.
acknowledges the Australian Research Council for financial
support. read less USED (high confidence) W. Zhu, G. Zheng, S. Cao, and H. He, “Thermal conductivity of amorphous SiO2 thin film: A molecular dynamics study,” Scientific Reports. 2018. link Times cited: 82 USED (high confidence) R. E. Jones, J. Rimsza, and L. Criscenti, “An atomic-scale evaluation of the fracture toughness of silica glass,” Journal of Physics: Condensed Matter. 2018. link Times cited: 4 Abstract: Using an atomistic technique consistent with continuum balan… read moreAbstract: Using an atomistic technique consistent with continuum balance laws and drawing on classical fracture mechanics theory, we estimate the resistance to fracture propagation of amorphous silica. We discuss correspondence and deviations from classical linear elastic fracture mechanics theory including size dependence, rigid/floppy modes of deformation, and the effects of surface energy and stress. read less USED (high confidence) C. Xiao, H. He, J. Li, and W. Zhu, “Kapitza resistance for nanoscale crystalline and amorphous silicon carbide,” 2018 19th International Conference on Thermal, Mechanical and Multi-Physics Simulation and Experiments in Microelectronics and Microsystems (EuroSimE). 2018. link Times cited: 0 Abstract: The interface between nanoscale films plays a very important… read moreAbstract: The interface between nanoscale films plays a very important role in semiconductor industry. In this paper, the interfacial thermal resistance (Kapitza resistance) of a crystalline and amorphous silicon carbide (SiC) heterojunction has been investigated by using molecular dynamics simulations. It is found that Kapitza resistance at crystalline and amorphous SiC interface depends on the interfacial coupling strength remarkably. Kapitza resistance in the strong interfacial coupling is significantly lower than that in weak coupling. The thickness of the heterojunction and temperature dependence of Kapitza resistance have also been examined. The results have shown that the Kapitza resistance decreases monotonically with the increase of temperature (from 300K to 800K). Moreover, Kapitza resistance can be effectively tuned by cross-plane strain. A 5% compressive strain is able to reduce the Kapitza resistance by 380% in weak coupling case. In contrast, a 5% tensile strain can increase Kapitza resistance by 13%. Our study provides useful guidance to the thermal management and heat dissipation across nanoscale crystalline and amorphous silicon carbide interface, in particular, for the design of silicon carbide nanowire based nano electronics devices. read less USED (high confidence) Z. Wang et al., “Abnormal separation of the silicon-oxygen bond in the liquid layering transition of silicon dioxide in a nanoslit.,” Physical chemistry chemical physics : PCCP. 2018. link Times cited: 0 Abstract: We investigated the unusual layering transition (LT) in quas… read moreAbstract: We investigated the unusual layering transition (LT) in quasi-2D liquid silicon dioxide (SiO2) confined in a nanoslit. Our results indicate that the slit size and pressure induce the abnormal LT in liquid SiO2, accompanied by a rapid change in the density, diffusion coefficient, pair correlation function and average potential energy. The silicon and oxygen atoms are almost completely separated under the extremely strong confinement effect, which is the characteristic feature of the LT. The negative slope of the LT lines in the phase diagram at different pressures suggests that a confinement-induced LT occurs at high pressure and a pressure-induced LT occurs at low pressure. read less USED (high confidence) J. Martínez-González, N. J. English, and A. Gowen, “Understanding the interface between silicon-based materials and water: Molecular-dynamics exploration of infrared spectra,” AIP Advances. 2017. link Times cited: 14 Abstract: Molecular-dynamics simulations for silicon, hydrogen- and hy… read moreAbstract: Molecular-dynamics simulations for silicon, hydrogen- and hydroxyl-terminated silicon in contact with liquid water, at 220 and 300 K, display water-density ‘ordering’ along the laboratory z-axis, emphasising the hydrophobicity of the different systems and the position of this first adsorbed layer. Density of states (DOS) of the oxygen and proton velocity correlation functions (VACFs) and infrared (IR) spectra of the first monolayer of adsorbed water, calculated via Fourier transformation, indicate similarities to more confined, ice-like dynamical behaviour (redolent of ice). It was observed that good qualitative agreement is obtained between the DOS for this first layer in all systems. The DOS for the lower-frequency zone indicates that for the interface studied (i.e., the first layer near the surface), the water molecules try to organise in a similar form, and that this form is intermediate between liquid water and ice. For IR spectra, scrutiny of the position of the highest-intensity peaks for the stret... read less USED (high confidence) L. M. Sandonas, G. Cuba-Supanta, R. Gutierrez, A. Dianat, C. Landauro, and G. Cuniberti, “Enhancement of thermal transport properties of asymmetric Graphene/hBN nanoribbon heterojunctions by substrate engineering,” Carbon. 2017. link Times cited: 23 USED (high confidence) N. Liao, B. Zheng, M. Zhang, and W. Xue, “First-principles calculation of lithium insertion into homogeneous a-SiC2/5O6/5 as high performance anode,” RSC Advances. 2017. link Times cited: 0 Abstract: Amorphous silicon oxycarbide is considered as a promising an… read moreAbstract: Amorphous silicon oxycarbide is considered as a promising anode material for new generation of lithium-ion batteries, and figuring out the lithiation mechanism is crucial for its application. In this work, first principle calculations are performed to study the atomic structures, formation energy and lithiation voltage of homogeneous SiC2/5O6/5. The interpretation of radial distribution, angular distribution and coordinate number suggests that the Si–O bond tends to break and the Li2O will form at the beginning of lithiation, then the LixO and the LiySi form with increasing Li concentration, which makes a major contribution to the capacity of SiC2/5O6/5. By the Li content dependence of the formation energies curve, the theoretical specific capacity of SiC2/5O6/5 is predicted as 1415 mA h g−1, which is comparable to the reversible capacity of 900 mA h g−1 in experiments. Both the formation energies and the voltage curves suggest lithium is preferable in incorporation with SiC2/5O6/5, and this is attributed to the formation of LixO and LiySi. read less USED (high confidence) M. Zhang, N. Liao, W. Xue, and P. Yang, “Large-scale molecular dynamics modeling of boron-doped amorphous SiCO ceramics,” Journal of Molecular Modeling. 2017. link Times cited: 4 USED (high confidence) W.-L. Lv and A. Henry, “Examining the Validity of the Phonon Gas Model in Amorphous Materials,” Scientific Reports. 2016. link Times cited: 50 USED (high confidence) S. Zhang et al., “Response to Extreme Temperatures of Mesoporous Silica MCM-41: Porous Structure Transformation Simulation and Modification of Gas Adsorption Properties.,” Langmuir : the ACS journal of surfaces and colloids. 2016. link Times cited: 12 Abstract: Molecular dynamics (MD) and Monte Carlo (MC) simulations wer… read moreAbstract: Molecular dynamics (MD) and Monte Carlo (MC) simulations were applied together for the first time to reveal the porous structure transformation mechanisms of mesoporous silica MCM-41 subjected to temperatures up to 2885 K. Silica was experimentally characterized to inform the models and enable prediction of changes in gas adsorption/separation properties. MD simulations suggest that the pore closure process is activated by a collective diffusion of matrix atoms into the porous region, accompanied by bond reformation at the surface. Degradation is kinetically limited, such that complete pore closure is postponed at high heating rates. We experimentally observe decreased gas adsorption with increasing temperature in mesoporous silica heated at fixed rates, due to pore closure and structural degradation consistent with simulation predictions. Applying the Kissinger equation, we find a strong correlation between the simulated pore collapse temperatures and the experimental values which implies an activation energy of 416 ± 17 kJ/mol for pore closure. MC simulations give the adsorption and selectivity for thermally treated MCM-41, for N2, Ar, Kr, and Xe at room temperature within the 1-10 000 kPa pressure range. Relative to pristine MCM-41, we observe that increased surface roughness due to decreasing pore size amplifies the difference of the absolute adsorption amount differently for different adsorbate molecules. In particular, we find that adsorption of strongly interacting molecules can be enhanced in the low-pressure region while adsorption of weakly interacting molecules is inhibited. This then results in higher selectivity in binary mixture adsorption in mesoporous silica. read less USED (high confidence) C. Melis, S. Giordano, L. Colombo, and G. Mana, “Density functional theory calculations of the stress of oxidised (1 1 0) silicon surfaces,” Metrologia. 2016. link Times cited: 9 Abstract: The measurement of the lattice-parameter of silicon by x-ray… read moreAbstract: The measurement of the lattice-parameter of silicon by x-ray interferometry assumes the use of strain-free crystals. This might not be the case because surface relaxation, reconstruction, and oxidation cause strains without the application of any external force. In a previous work, this intrinsic strain was estimated by a finite element analysis, where the surface stress was modeled by an elastic membrane having a 1 N m−1 tensile strength. The present paper quantifies the surface stress by a density functional theory calculation. We found a value exceeding the nominal value used, which potentially affects the measurement accuracy. read less USED (high confidence) N. Liao, B. Zheng, M. Zhang, and W. Xue, “Atomic investigation on reversible lithium storage in amorphous silicon oxycarbide as a high power anode material,” Journal of Materials Chemistry. 2016. link Times cited: 50 Abstract: Silicon oxycarbide (SiCO) has a remarkable reversible capaci… read moreAbstract: Silicon oxycarbide (SiCO) has a remarkable reversible capacity of lithium and is believed to be a promising anode material for the new generation of lithium-ion batteries. Although current experiments have provided some information on lithium storage in SiCO, further study on the origin of reversible capacity needs to be conducted at the atomic scale. In this work, first principles calculations are used to investigate reversible lithium storage in five SiCO structures with different compositions. Based on lithiated structures, the Si–O bond tends to break and Li2O forms at the beginning of lithiation and then LixO and LiySi form with increasing Li concentration, which make a major contribution to the Li capacity. The carbon atoms do not attract lithium but form a stable C–C domain to maintain the stability of the lithiated system; this is also verified by the root mean-square deviation of C. The free volume of the structures tends to decrease with increasing carbon content, implying that the void is not the major resource for lithium storage. Stoichiometric glass without free carbon presents very low reversible capacity. The reversible capacity tends to increase with higher carbon concentration; however, it would reach a maximum value and begin to decrease when the carbon content increases further. read less USED (high confidence) H. Seyf and A. Henry, “A method for distinguishing between propagons, diffusions, and locons,” Journal of Applied Physics. 2016. link Times cited: 81 Abstract: The majority of intuition on phonon transport has been deriv… read moreAbstract: The majority of intuition on phonon transport has been derived from studies of homogenous crystalline solids, where the atomic composition and structure are periodic. For this specific class of materials, the solutions to the equations of motions for the atoms (in the harmonic limit) result in plane wave modulated velocity fields for the normal modes of vibration. However, it has been known for several decades that whenever a system lacks periodicity, either compositional or structural, the normal modes of vibration can still be determined (in the harmonic limit), but the solutions take on different characteristics and many modes may not be plane wave modulated. Previous work has classified the types of vibrations into three primary categories, namely, propagons, diffusions, and locons. One can use the participation ratio to distinguish locons, from propagons and diffusons, which measures the extent to which a mode is localized. However, distinguishing between propagons and diffusons has remained a challe... read less USED (high confidence) Z. Liang and P. Keblinski, “Sound attenuation in amorphous silica at frequencies near the boson peak,” Physical Review B. 2016. link Times cited: 9 USED (high confidence) C. Y. Chuang, L. Zepeda-Ruiz, S. Han, and T. Sinno, “Direct molecular dynamics simulation of Ge deposition on amorphous SiO2 at experimentally relevant conditions,” Surface Science. 2015. link Times cited: 1 USED (high confidence) J. Zhang, Y. Hong, Z. Tong, Z. Xiao, H. Bao, and Y. Yue, “Molecular dynamics study of interfacial thermal transport between silicene and substrates.,” Physical chemistry chemical physics : PCCP. 2015. link Times cited: 41 Abstract: In this work, the interfacial thermal transport across silic… read moreAbstract: In this work, the interfacial thermal transport across silicene and various substrates, i.e., crystalline silicon (c-Si), amorphous silicon (a-Si), crystalline silica (c-SiO2) and amorphous silica (a-SiO2) are explored by classical molecular dynamics (MD) simulations. A transient pulsed heating technique is applied in this work to characterize the interfacial thermal resistance in all hybrid systems. It is reported that the interfacial thermal resistances between silicene and all substrates decrease nearly 40% with temperature from 100 K to 400 K, which is due to the enhanced phonon couplings from the anharmonicity effect. Analysis of phonon power spectra of all systems is performed to interpret simulation results. Contradictory to the traditional thought that amorphous structures tend to have poor thermal transport capabilities due to the disordered atomic configurations, it is calculated that amorphous silicon and silica substrates facilitate the interfacial thermal transport compared with their crystalline structures. Besides, the coupling effect from substrates can improve the interface thermal transport up to 43.5% for coupling strengths χ from 1.0 to 2.0. Our results provide fundamental knowledge and rational guidelines for the design and development of the next-generation silicene-based nanoelectronics and thermal interface materials. read less USED (high confidence) W. Gao, K. Liechti, and R. Huang, “Wet adhesion of graphene,” Extreme Mechanics Letters. 2015. link Times cited: 18 USED (high confidence) W. Li, X. Wang, X. Zhang, S. Zhao, H. Duan, and J. Xue, “Mechanism of the Defect Formation in Supported Graphene by Energetic Heavy Ion Irradiation: the Substrate Effect,” Scientific Reports. 2015. link Times cited: 72 USED (high confidence) S. Neogi et al., “Tuning thermal transport in ultrathin silicon membranes by surface nanoscale engineering.,” ACS nano. 2015. link Times cited: 105 Abstract: A detailed understanding of the connections of fabrication a… read moreAbstract: A detailed understanding of the connections of fabrication and processing to structural and thermal properties of low-dimensional nanostructures is essential to design materials and devices for phononics, nanoscale thermal management, and thermoelectric applications. Silicon provides an ideal platform to study the relations between structure and heat transport since its thermal conductivity can be tuned over 2 orders of magnitude by nanostructuring. Combining realistic atomistic modeling and experiments, we unravel the origin of the thermal conductivity reduction in ultrathin suspended silicon membranes, down to a thickness of 4 nm. Heat transport is mostly controlled by surface scattering: rough layers of native oxide at surfaces limit the mean free path of thermal phonons below 100 nm. Removing the oxide layers by chemical processing allows us to tune the thermal conductivity over 1 order of magnitude. Our results guide materials design for future phononic applications, setting the length scale at which nanostructuring affects thermal phonons most effectively. read less USED (high confidence) L. Chen and S. Kumar, “Heat Dissipation Mechanism at Carbon Nanotube Junctions on Silicon Oxide Substrate,” Journal of Heat Transfer-transactions of The Asme. 2014. link Times cited: 7 Abstract: This study investigates heat dissipation at carbon nanotube … read moreAbstract: This study investigates heat dissipation at carbon nanotube (CNT) junctions supported on silicon dioxide substrate using molecular dynamics simulations. The temperature rise in a CNT (~top CNT) not making direct contact with the oxide substrate but only supported by other CNTs (~bottom CNT) is observed to be hundreds of degree higher compared to the CNTs well-contacted with the substrate at similar power densities. The analysis of spectral temperature decay of CNT-oxide system shows very fast intra-tube energy transfer in a CNT from high frequency band to intermediate frequency bands. The low frequency phonon band (0-5 THz) of top CNT shows two-stage energy relaxation which results from the efficient coupling of low frequency phonons in the CNT-oxide system and the blocking of direct transport of high and intermediate frequency phonons of top CNT to the oxide substrate by bottom CNT. read less USED (high confidence) A. Galashev, “Computer study of the Raman spectra and infrared optical properties of gallium nitride and gallium arsenic nanoparticles with SiO2 core and shell,” Journal of Nanoparticle Research. 2014. link Times cited: 2 USED (high confidence) B. Kang, S. D. Nath, H. Kim, and S.-G. Kim, “Parallel laser fabrication of film-embedded microstructures using reusable functionalized template,” Journal of Applied Physics. 2014. link Times cited: 2 Abstract: This study proposes a new parallel mass-production method fo… read moreAbstract: This study proposes a new parallel mass-production method for obtaining microstructures embedded in flexible films, utilizing a laser-processed reusable functionalized template and a laser-induced adhesive transfer. This physical shape-free template can be cost-effectively fabricated by means of the laser-induced plasmonic defunctionalization of a self-assembled monolayer. The resulting metal nanoparticle microstructure, deposited self-selectively on the template, is transferred to a flexible film by a photo-induced instantaneous interfacial adhesion film in parallel; this process being optimized using molecular dynamics simulations. This method is demonstrated to be capable of the high-efficiency and eco-friendly production of high resolution and durable microstructures in flexible films, using a reusable template to eliminate material waste. Moreover, key design parameters such as the resolution, thickness, type, and shape of microstructures can be actively changed. read less USED (high confidence) A. Galashev, “Computer study of the spectral characteristics and structures of (GaN)54(SiO2)50 nanoparticles,” Journal of Surface Investigation. X-ray, Synchrotron and Neutron Techniques. 2013. link Times cited: 2 USED (high confidence) N. Liao, W. Xue, H. Zhou, and M. Zhang, “Investigation on high temperature fracture properties of amorphous silicon dioxide by large-scale atomistic simulations,” Journal of Materials Science: Materials in Electronics. 2013. link Times cited: 4 USED (high confidence) M. Gupta, L. Chen, D. Estrada, A. Behnam, E. Pop, and S. Kumar, “Impact of thermal boundary conductances on power dissipation and electrical breakdown of carbon nanotube network transistors,” Journal of Applied Physics. 2012. link Times cited: 19 Abstract: estimate the average CNT-SiO2 TBC as g � 0.16Wm � 1 K � 1 an… read moreAbstract: estimate the average CNT-SiO2 TBC as g � 0.16Wm � 1 K � 1 and the TBC at CNT junctions as GC � 2.4 pWK � 1 . We find the peak power dissipation in CN-TFTs is more strongly correlated to the TBC of the CNT-substrate interface than to the TBC at CNT junctions. Molecular dynamics simulations of crossed CNT junctions also reveal that the top CNT is buckled over � 30nm lengths, losing direct contact with the substrate and creating highly localized hot-spots. Our results provide new insights into CNT network properties which can be engineered to enhance performance of CN-TFTs for macro and flexible electronics applications. V C 2012 American read less USED (high confidence) J. Chen, G. Zhang, and B. Li, “Thermal Contact Resistance Across Nanoscale Silicon Dioxide and Silicon Interface,” arXiv: Materials Science. 2012. link Times cited: 123 Abstract: Silicon dioxide and silicon (SiO$_{2}$/Si) interface plays a… read moreAbstract: Silicon dioxide and silicon (SiO$_{2}$/Si) interface plays a very important role in semiconductor industry. However, at nanoscale, its interfacial thermal properties haven't been well understood so far. In this paper, we systematically study the interfacial thermal resistance (Kapitza resistance) of a heterojunction composed of amorphous silicon dioxide and crystalline silicon by using molecular dynamics simulations. Numerical results have shown that Kapitza resistance at SiO$_{2}$/Si interface depends on the interfacial coupling strength remarkably. In the weak interfacial coupling limit, Kapitza resistance depends on both the detailed interfacial structure and the length of the heterojunction, showing large fluctuation among different samples. In contrast, it is almost insensitive to the detailed interfacial structure or the length of the heterojunction in the strong interfacial coupling limit, giving rise to a nearly constant value around 0.9 $\times10^{-9}$ m$^{2}$KW$^{-1}$ at room temperature. Moreover, the temperature dependent Kapitza resistance in the strong interfacial coupling limit has also been examined. Our study provides useful guidance to the thermal management and heat dissipation across nanoscale SiO$_{2}$/Si interface, in particular for the design of silicon nanowire based nano electronics and photonics devices. read less USED (high confidence) J. Fang and L. Pilon, “Tuning thermal conductivity of nanoporous crystalline silicon by surface passivation: A molecular dynamics study,” Applied Physics Letters. 2012. link Times cited: 18 Abstract: Surface passivation of nanoporous crystalline silicon can re… read moreAbstract: Surface passivation of nanoporous crystalline silicon can reduce its thermal conductivity. This was established using equilibrium molecular dynamics simulations. The porosity varied from 8% to 38% while the pore diameter ranged from 1.74 to 2.93 nm. Hydrogen and oxygen passivation reduced thermal conductivity by 11% to 17% and 37% to 51% depending on porosity at 500 K, respectively. The hydrogen passivation effect decreased with increasing temperature. Vibrational spectra of oxygen overlapped with those of silicon at low frequencies. Therefore, oxygen passivation enhanced phonon scattering at solid matrix boundaries, resulting in stronger thermal conductivity reduction than that caused by hydrogen passivation. read less USED (high confidence) E. Lampin, Q. Nguyen, P. A. Francioso, and F. Cleri, “Thermal boundary resistance at silicon-silica interfaces by molecular dynamics simulations,” Applied Physics Letters. 2012. link Times cited: 61 Abstract: We use molecular dynamics simulations to study the heat tran… read moreAbstract: We use molecular dynamics simulations to study the heat transfer at the interface between crystalline Si and amorphous silica. In order to quantify the thermal boundary resistance, we compare the results of two simulation methods: one in which we apply a stationary thermal gradient across the interface, trying to extract the thermal resistance from the temperature jump; the other based on the exponential approach to thermal equilibrium, by monitoring the relaxation times of the heat flux exchanged across the interface. We compare crystalline Si/amorphous Si vs. crystalline Si/amorphous silica interfaces to assess the relative importance of structural disordering vs. chemistry difference. read less USED (high confidence) E. Lampin and C. Krzeminski, “Regrowth of oxide-embedded amorphous silicon studied with molecular dynamics,” Journal of Applied Physics. 2011. link Times cited: 11 Abstract: Classical molecular dynamics simulations are applied to the … read moreAbstract: Classical molecular dynamics simulations are applied to the study of amorphous silicon regrowth in a nanodevice. A simplified atomistic amorphous nanostructure presenting the main features of a FinFET device is designed. A thermal treatment is used to simulate the annealing of the atomic model. The structure after annealing is very close to what observed experimentally, with perfect crystal near the silicon seed, an intermediate crystalline layer presenting [111] twins, and an upper terminal region of polysilicon. The comparison with 2D system suggests surface proximity effects that impact the probability to form grains and twins. As a consequence, it seems like the solid phase epitaxy was arrested in the nanostructure. read less USED (high confidence) R. Renou et al., “Investigating ramp wave propagation inside silica glass with laser experiments and molecular simulations.” 2018. link Times cited: 2 Abstract: Under elastic shock compression silica glass exhibits a very… read moreAbstract: Under elastic shock compression silica glass exhibits a very specific behaviour. A shock propagating inside a material is usually seen as the propagation of a discontinuity. However in silica glass, shocks are unstable and lead to the propagation of a ramp wave where the shock front becomes gradually larger over time. Ramp waves were already reported in the literature, however their origin remain uncertain. This work presents an original study combining laser shock-induced experiments and molecular dynamics simulation aiming to improve the understanding of the mechanisms involved. Experimental ramp waves were directly observed using shadowgraphy technique allowing for an estimation of the head and tail velocities. Molecular dynamics simulations were carried out in order to reproduce ramp waves and to gain insight into the material properties. Ramp waves were observed for both elastic and plastic shockwaves. In the latter case, the plastic waves were preceded by an elastic ramp precursor. The sound speed, related to the material compressibility, was found to decrease with increasing pressure, as observed experimentally for quasi-static hydrostatic loading, thus providing an explanation for the instabilities that lead to the propagation of ramp waves. read less USED (high confidence) X. Hu, C. Tourek, Z. Ye, S. Sundararajan, and A. Martini, “Structural and Chemical Evolution of the Near-Apex Region of an Atomic Force Microscope Tip Subject to Sliding,” Tribology Letters. 2013. link Times cited: 12 USED (low confidence) C. Y. Zhao, C. Yang, Y. B. Tao, and Y. L. He, “Interfacial nanolayer effect on thermophysical properties of silica-paraffin phase change material - A molecular dynamics simulation,” International Journal of Heat and Mass Transfer. 2024. link Times cited: 0 USED (low confidence) X. Zhou, M. Hou, R. Liu, and B. Liu, “Fabrication of beryllium oxide based fully ceramic microencapsulated nuclear fuels with dispersed TRISO particles by pressureless sintering method,” Journal of Nuclear Materials. 2024. link Times cited: 0 USED (low confidence) Y.-B. Jiao et al., “Threshold displacement energy of amorphous SiO2: A molecular dynamics study,” Journal of Non-Crystalline Solids. 2023. link Times cited: 0 USED (low confidence) I. Nikolaev, P. Stishenko, V. V. Yakovlev, and N. Korobeishchikov, “Effect of gas cluster species on crater formation for fused silica,” Journal of Non-Crystalline Solids. 2023. link Times cited: 0 USED (low confidence) S. Uchida, K. Fujiwara, and M. Shibahara, “Microscopic properties of forces from ice solidification interface acting on silica surfaces based on molecular dynamics simulations.,” Physical chemistry chemical physics : PCCP. 2023. link Times cited: 0 Abstract: The origin of the forces acting on a silica surface from an … read moreAbstract: The origin of the forces acting on a silica surface from an ice solidification interface was investigated to understand the solidification phenomenon and its impact on nanometer-scale structures using molecular dynamics simulations. The microscopic forces were determined by appropriately averaging the forces acting on the silica wall from the water molecules in time and space; the time evolutions of these microscopic forces during the solidification processes were investigated for three types of silica surfaces. The results indicate that the microscopic forces fluctuate more after the solidification interface makes contact with the wall surface. To visualize the changes in the microscopic forces and hydrogen bonds due to solidification, their differences compared to the liquid state were calculated. When the solidification interface is near the wall, the changes in these microscopic forces and hydrogen bonds due to solidification are correlated. This tendency is more significant for an amorphous wall and a wall with a structure than for a crystalline wall. The changes in the microscopic force depend on the water molecules that behave as acceptors when forming the hydrogen bonds with the wall and on the configuration of the silanol groups on the silica surfaces. read less USED (low confidence) C. Zhang, Y. Pan, Y. Bi, and X. Cao, “Fracture behavior and energy efficiency of silica under a tensile load using molecular dynamics,” Engineering Fracture Mechanics. 2023. link Times cited: 0 USED (low confidence) S. Ke et al., “Simulation of Crystal Nuclei at the Liquid-Air Interface toward Morphology Control via Surface Tension,” The Journal of Physical Chemistry C. 2023. link Times cited: 0 USED (low confidence) S. Thakur and A. Giri, “Role of Anharmonicity in Dictating the Thermal Boundary Conductance across Interfaces Comprised of Two-Dimensional Materials,” Physical Review Applied. 2023. link Times cited: 0 USED (low confidence) W. Yue, T. Luo, and K. Liu, “Trade-Off Between Permeability and Compressive Strength for Aerated Concrete-Based Material with Fly-Ash Under High Pressure,” Transport in Porous Media. 2023. link Times cited: 0 USED (low confidence) L. Gao, Y. Sun, and H.-B. Yu, “Mobility percolation as a source of Johari-Goldstein relaxation in glasses,” Physical Review B. 2023. link Times cited: 1 USED (low confidence) X. Ma, X. Kang, and J. Cao, “Origin of the elastic anisotropy of silica particles: Insights from first-principles calculations and nanoindentation molecular dynamic simulations,” Computers and Geotechnics. 2023. link Times cited: 1 USED (low confidence) K. Huang, W. Wu, S. Xu, P. Yan, Z. Wei, and Q. Xu, “CO2‐Induced Modulation of Si–O Bonds for Low Temperature Plastic Deformation of Amorphous Silica Nanoparticles with Enhanced Photoluminescence,” ENERGY & ENVIRONMENTAL MATERIALS. 2023. link Times cited: 0 USED (low confidence) “Subcontinuum scale analysis of diamond lattice films through spatial multi-level coarsening method,” Thin-Walled Structures. 2023. link Times cited: 2 USED (low confidence) J. Zhou, Y. Lu, C. Wang, D. Feng, H. Zhang, and Y. Li, “Molecular dynamics study of graphene-coated reinforced tribomechanical properties: Hard versus soft substrates,” Computational Materials Science. 2023. link Times cited: 1 USED (low confidence) J. Wu, Z. Rui, and Y. Dong, “Effect of substrate temperature on adhesion at liquid-aluminum/silica interface by phantom wall method,” Materials Today Communications. 2023. link Times cited: 0 USED (low confidence) M. Tahani, E. Postek, and T. Sadowski, “Diffusion and Interdiffusion Study at Al- and O-Terminated Al2O3/AlSi12 Interface Using Molecular Dynamics Simulations,” Materials. 2023. link Times cited: 2 Abstract: The equivalent characteristics of the materials’ interfaces … read moreAbstract: The equivalent characteristics of the materials’ interfaces are known to impact the overall mechanical properties of ceramic–metal composites significantly. One technological method that has been suggested is raising the temperature of the liquid metal to improve the weak wettability of ceramic particles with liquid metals. Therefore, as the first step, it is necessary to produce the diffusion zone at the interface by heating the system and maintaining it at a preset temperature to develop the cohesive zone model of the interface using mode I and mode II fracture tests. This study uses the molecular dynamics method to study the interdiffusion at the interface of α-Al2O3/AlSi12. The hexagonal crystal structure of aluminum oxide with the Al- and O-terminated interfaces with AlSi12 are considered. A single diffusion couple is used for each system to determine the average main and cross ternary interdiffusion coefficients. In addition, the effect of temperature and the termination type on the interdiffusion coefficients is examined. The results demonstrate that the thickness of the interdiffusion zone is proportional to the annealing temperature and time, and Al- and O-terminated interfaces exhibit similar interdiffusion properties. read less USED (low confidence) A. Mysovsky and A. Paklin, “Molecular Dynamics Modeling of SiO2 Melts and Glass Formation Processes,” Glass Physics and Chemistry. 2023. link Times cited: 0 USED (low confidence) M. Liao, P. Nicolini, and T. Polcar, “Separating anisotropic and isotropic friction between atomic force microscope tips and atomically flat surfaces,” Physical Review B. 2023. link Times cited: 0 USED (low confidence) F. Grigoriev and V. Sulimov, “Atomistic Simulation of Physical Vapor Deposition of Optical Thin Films,” Nanomaterials. 2023. link Times cited: 0 Abstract: A review of the methods and results of atomistic modeling of… read moreAbstract: A review of the methods and results of atomistic modeling of the deposition of thin optical films and a calculation of their characteristics is presented. The simulation of various processes in a vacuum chamber, including target sputtering and the formation of film layers, is considered. Methods for calculating the structural, mechanical, optical, and electronic properties of thin optical films and film-forming materials are discussed. The application of these methods to studying the dependences of the characteristics of thin optical films on the main deposition parameters is considered. The simulation results are compared with experimental data. read less USED (low confidence) A. Leino, V. Jantunen, P. Mota‐Santiago, P. Kluth, and F. Djurabekova, “Insights into nanoparticle shape transformation by energetic ions.,” Scientific reports. 2023. link Times cited: 0 USED (low confidence) J. Chen, L. Fang, H. Chen, K. Sun, S. Dang, and J. Han, “Soft abrasive facilitating materials removal of SiO2/Si bilayer materials: A molecular dynamics study,” Materials Chemistry and Physics. 2023. link Times cited: 2 USED (low confidence) L. Wang, “The effect of substrate vibration on Ag nanoparticle formation on SiO2 via thermally-induced dewetting: a molecular dynamics study,” Thin Solid Films. 2023. link Times cited: 0 USED (low confidence) S. S. Moosavi and A. R. Zolghadr, “Structural Transitions of Anionic, Cationic, and Nonionic Surfactant Solutions Confined between Amorphous SiO2 Slabs: Molecular Dynamics Simulations,” Industrial & Engineering Chemistry Research. 2022. link Times cited: 1 USED (low confidence) M. Y. Yang, G. Tang, Q. Sheng, L. Guo, and H. Zhang, “Atomic-level sintering mechanism of silica aerogels at high temperatures: structure evolution and solid thermal conductivity,” International Journal of Heat and Mass Transfer. 2022. link Times cited: 4 USED (low confidence) N. Anbarloui and E. Nadimi, “Different forms of silicon nanotubes and their interactions with DNA nucleotides: A molecular dynamics study,” Physica B: Condensed Matter. 2022. link Times cited: 1 USED (low confidence) R. Alfaridzi, M. L. Nietiadi, H. Urbassek, and Y. Rosandi, “The effect of enclosed water-ice pockets on porous silica cluster collisions,” Icarus. 2022. link Times cited: 1 USED (low confidence) Y. Zhang et al., “Simultaneous electrical and thermal rectification in a monolayer lateral heterojunction,” Science. 2022. link Times cited: 24 Abstract: Efficient waste heat dissipation has become increasingly cha… read moreAbstract: Efficient waste heat dissipation has become increasingly challenging as transistor size has decreased to nanometers. As governed by universal Umklapp phonon scattering, the thermal conductivity of semiconductors decreases at higher temperatures and causes heat transfer deterioration under high-power conditions. In this study, we realized simultaneous electrical and thermal rectification (TR) in a monolayer MoSe2-WSe2 lateral heterostructure. The atomically thin MoSe2-WSe2 heterojunction forms an electrical diode with a high ON/OFF ratio up to 104. Meanwhile, a preferred heat dissipation channel was formed from MoSe2 to WSe2 in the ON state of the heterojunction diode at high bias voltage with a TR factor as high as 96%. Higher thermal conductivity was achieved at higher temperatures owing to the TR effect caused by the local temperature gradient. Furthermore, the TR factor could be regulated from maximum to zero by rotating the angle of the monolayer heterojunction interface. This result opens a path for designing novel nanoelectronic devices with enhanced thermal dissipation. Description A one-way street for heat For most materials, reversing the direction of the thermal gradient does not change the thermal conductivity. The difficulty in finding thermal rectifiers impedes the development of thermal diodes that would be important for managing heat flow. Y. Zhang et al. found that a heterostructure made of molybdenum diselenide and tungsten diselenide rectifies both electricity and heat. The magnitude of the effect depends on the relative geometry of the interface and the thermal gradient. The discovery provides the opportunity to develop more effective heat dissipation in highly integrated circuits. —BG A monolayer heterostructure rectifier was developed to obtain simultaneous electrical and thermal rectification. read less USED (low confidence) N. G. Korobeishchikov, P. Stishenko, I. Nikolaev, and V. Yakovlev, “Silica sputtering by noble gas projectiles: elucidating the effect of cluster species with molecular dynamic simulation,” Plasma Chemistry and Plasma Processing. 2022. link Times cited: 1 USED (low confidence) L. Wang, “Dewetting of ultrathin Ag film with random vacancy defects on a SiO2 substrate: a molecular dynamics simulation,” RSC Advances. 2022. link Times cited: 0 Abstract: The spinodal instability and thermal nucleation mechanisms s… read moreAbstract: The spinodal instability and thermal nucleation mechanisms successfully describe the dewetting of metallic thin films. The previous research mainly focuses on homogeneous and continuous films. However, less attention is paid to the effect of random vacancy defects that frequently appear in actual situations on the film dewetting. In this work, the thermally-induced dewetting of a 0.4 nm thick ultrathin Ag film with different vacancy rate (f) ranging from 0.01 to 0.5 on a SiO2 substrate is investigated by the molecular dynamics (MD) simulation. Thermal nucleation and growth of holes appear in the dewetting process. The characteristic dewetting time (t) decreases dramatically with the increase of vacancy rate (f) of the Ag film. This is possibly because the presence of vacancy defects effectively reduce the incubation period of the initial holes, which is significant even for a very small vacancy rate less than 0.05. read less USED (low confidence) Y. Du et al., “Comparison of glancing-angle scatterings on different materials in a high aspect ratio plasma etching process using molecular dynamics simulation,” Journal of Vacuum Science & Technology A. 2022. link Times cited: 3 Abstract: In plasma etching for microelectronics fabrication, one of t… read moreAbstract: In plasma etching for microelectronics fabrication, one of the objectives is to produce a high aspect ratio (HAR) via and trench structures. A principal contributor to the HAR feature shape is the manner in which energetic ions interact with sidewalls inside the feature. The scattering angle and energy loss of ions reflecting from sidewalls determine the sidewall slope and can lead to defects such as microtrenching and bowing. Understanding how ions interact with sidewalls can improve our control of the critical dimensions of HAR features. Ions accelerated in the plasma sheath arrive in the feature with energies as large as a few keV and initially strike the sidewalls at glancing angles. These scattering events extend to the photolithographic mask. Scattering from the mask at glancing angles can produce ions incident into the underlying feature with a broader angular distribution, leading to less desirable feature properties. In this work, results are discussed from Molecular Dynamics (MD) simulations of glancing-angle scattering of argon ions from three materials common to HAR etch: polystyrene (as a photoresist surrogate), amorphous carbon (a hard mask material), and [Formula: see text] (a common insulating material used in microelectronics devices). Results from simulations reveal a transition from specular scattering to diffuse scattering as the angle of the incident ion decreases (90[Formula: see text] being glancing incidence) and incident energy increases. Scattering from polystyrene is more diffuse compared to amorphous carbon and [Formula: see text] for identical incident ion conditions. read less USED (low confidence) Y. Lin et al., “Interfacial mechanical properties of tetrahydrofuran hydrate-solid surfaces: Implications for hydrate management.,” Journal of colloid and interface science. 2022. link Times cited: 9 USED (low confidence) Y. Hu, K. Jiang, K. Liew, and L.-W. Zhang, “Nanoarray-Embedded Hierarchical Surfaces for Highly Durable Dropwise Condensation,” Research. 2022. link Times cited: 2 Abstract: Durable dropwise condensation of saturated vapor is of signi… read moreAbstract: Durable dropwise condensation of saturated vapor is of significance for heat transfer and energy saving in extensive industrial applications. While numerous superhydrophobic surfaces can promote steam condensation, maintaining discrete microdroplets on surfaces without the formation of a flooded filmwise condensation at high subcooling remains challenging. Here, we report the development of carbon nanotube array-embedded hierarchical composite surfaces that enable ultra-durable dropwise condensation under a wide range of subcooling (ΔTsub = 8 K–38 K), which outperforms existing nanowire surfaces. This performance stems from the combined strategies of the hydrophobic nanostructures that allow efficient surface renewal and the patterned hydrophilic micro frames that protect the nanostructures and also accelerate droplet nucleation. The synergistic effects of the composite design ensure sustained Cassie wetting mode and capillarity-governed droplet mobility (Bond number < 0.055) as well as the large specific volume of condensed droplets, which contributes to the enhanced condensation heat transfer. Our design provides a feasible alternative for efficiently transferring heat in a vapor environment with relatively high temperatures through the tunable multiscale morphology. read less USED (low confidence) D. N. Trong, V. C. Long, Ș. Ţălu, U. Saraç, P. N. Dang, and K. P. Huu, “A New Study on the Structure, and Phase Transition Temperature of Bulk Silicate Materials by Simulation Method of Molecular Dynamics,” Journal of Composites Science. 2022. link Times cited: 2 Abstract: In this paper, the structure and phase transition temperatur… read moreAbstract: In this paper, the structure and phase transition temperature of bulk silicate materials are studied by the simulation method (SM) of molecular dynamics (MD). In this research, all samples are prepared on the same nanoscale material model with the atomic number of 3000 atoms, for which the SM of MD is performed with Beest-Kramer-van Santen and van Santen pair interaction potentials under cyclic boundary conditions. The obtained results show that both the model size (l) and the total energy of the system (Etot) increase slowly in the low temperature (T) region (negative T values) at pressure (P), P = 0 GPa. However, the increase of l determines the Etot value with very large values in the high T region. It is found that l decreases greatly in the high T region with increasing P, and vice versa. In addition, when P increases, the decrease in the Etot value is small in the low T region, but large in the high T region. As a consequence, a change appears in the lengths of the Si-Si, Si-O, and O-O bonds, which are very large in the high T and high P regions, but insignificant in the low T and low P regions. Furthermore, the structural unit number of SiO7 appears at T > 2974 K in the high P region. The obtained results will serve as the basis for future experimental studies to exploit the stored energy used in semiconductor devices. read less USED (low confidence) M. Dung, “Structural properties of silica under the temperature,” Journal of Physics: Conference Series. 2022. link Times cited: 0 Abstract: The structure of the SiO2 system consisting of 12800 atoms i… read moreAbstract: The structure of the SiO2 system consisting of 12800 atoms is performed via molecular dynamics simulation with the Tersoff potential. Our simulation shows that the onset of the melting temperature is at 3450 K. This value is much higher than the previous result of Ringdalen and co-workers [26]. The structural evolution of the system is analyzed through the pair radial distribution function, the distribution of the bond length and the distribution of the bond angle. Structural parameters are compared with the previous experimental and simulated results. The mean square displacement is also discussed in this paper. read less USED (low confidence) B. He, T. Vo, and P. Newell, “Investigation of fracture in porous materials: a phase-field fracture study informed by ReaxFF,” Engineering with Computers. 2022. link Times cited: 4 USED (low confidence) J. Chen, L. Fang, H. Chen, K. Sun, and J. Han, “Indenter Size Effect on Stress Relaxation Behaviors of Surface-modified Silicon: A Molecular Dynamics Study,” Journal of Wuhan University of Technology-Mater. Sci. Ed. 2022. link Times cited: 0 USED (low confidence) J. Chen, L. Fang, H. Chen, K. Sun, S. Dang, and J. Han, “The loading speed facilitating stress relaxation behaviors of surface-modified silicon: a molecular dynamics study,” Journal of Molecular Modeling. 2022. link Times cited: 1 USED (low confidence) Y. Zhou et al., “Unusual Deformation and Fracture in Gallium Telluride Multilayers,” The Journal of Physical Chemistry Letters. 2022. link Times cited: 8 Abstract: The deformation and fracture mechanism of two-dimensional (2… read moreAbstract: The deformation and fracture mechanism of two-dimensional (2D) materials are still unclear and not thoroughly investigated. Given this, mechanical properties and mechanisms are explored on example of gallium telluride (GaTe), a promising 2D semiconductor with an ultrahigh photoresponsivity and a high flexibility. Hereby, the mechanical properties of both substrate-supported and suspended GaTe multilayers were investigated through Berkovich-tip nanoindentation instead of the commonly used AFM-based nanoindentation method. An unusual concurrence of multiple pop-in and load-drop events in loading curve was observed. Theoretical calculations unveiled this concurrence originating from the interlayer-sliding mediated layers-by-layers fracture mechanism in GaTe multilayers. The van der Waals force dominated interlayer interactions between GaTe and substrates was revealed much stronger than that between GaTe interlayers, resulting in the easy sliding and fracture of multilayers within GaTe. This work introduces new insights into the deformation and fracture of GaTe and other 2D materials in flexible electronics applications. read less USED (low confidence) A. Gabourie, Ç. Köroğlu, and E. Pop, “Substrate-dependence of monolayer MoS2 thermal conductivity and thermal boundary conductance,” Journal of Applied Physics. 2022. link Times cited: 10 Abstract: The thermal properties of two-dimensional (2D) materials, su… read moreAbstract: The thermal properties of two-dimensional (2D) materials, such as MoS2, are known to be affected by interactions with their environment, but this has primarily been studied only with SiO2 substrates. Here, we compare the thermal conductivity (TC) and thermal boundary conductance (TBC) of monolayer MoS2 on amorphous (a-) and crystalline (c-) SiO2, AlN, Al2O3, and h-BN monolayers using molecular dynamics. The room temperature, in-plane TC of MoS2 is ∼38 Wm−1 K−1 on amorphous substrates and up to ∼68 Wm−1 K−1 on crystalline substrates, with most of the difference due to substrate interactions with long-wavelength MoS2 phonons (<2 THz). An h-BN monolayer used as a buffer between MoS2 and the substrate causes the MoS2 TC to increase by up to 50%. Length-dependent calculations reveal TC size effects below ∼2 μm and show that the MoS2 TC is not substrate- but size-limited below ∼100 nm. We also find that the TBC of MoS2 with c-Al2O3 is over twice that with c-AlN despite a similar MoS2 TC on both, indicating that the TC and TBC could be tuned independently. Finally, we compare the thermal resistance of MoS2 transistors on all substrates and find that MoS2 TBC is the most important parameter for heat removal for long-channel (>150 nm) devices, while TBC and TC are equally important for short channels. This work provides important insights for electro-thermal applications of 2D materials on various substrates. read less USED (low confidence) Y. Zhou et al., “Investigation of Deformation and Fracture Mechanisms in Two-dimensional Gallium Telluride Multilayers Using Nanoindentation.” 2022. link Times cited: 0 Abstract: Two-dimensional (2D) materials possess great potential for f… read moreAbstract: Two-dimensional (2D) materials possess great potential for flexible devices, ascribing to their outstanding electrical, optical, and mechanical properties. However, their mechanical deformation property and fracture mechanism, which are inescapable in many applications like flexible optoelectronics, are still unclear or not thoroughly investigated due methodology limitations. In light of this, such mechanical properties and mechanisms are explored on example of gallium telluride (GaTe), a promising optoelectronic candidate with an ultrahigh photo-responsibility and a high plasticity within 2D family. Considering the driving force insufficient in atomic force microscopy (AFM)-based nanoindentation method, here the mechanical properties of both substrate-supported and suspended GaTe multilayers were systematically investigated through full-scale Berkovich-tip nanoindentation, micro-Raman spectroscopy, AFM, and scanning electron microscopy. An unusual concurrence of multiple pop-in and load-drop events in loading curve was observed. By further correlating to molecular dynamics calculations, this concurrence was unveiled originating from the interlayer sliding mediated layers-by-layers fracture mechanism within GaTe multilayers. The van der Waals force between GaTe multilayers and substrates was revealed much stronger than that between GaTe interlayers, resulting in the easy sliding and fracture of multilayers within GaTe. This work provides new insights into the deformation and fracture mechanisms of GaTe and other similar 2D multilayers in flexible applications. read less USED (low confidence) S. Paul, R. Torsi, J. Robinson, and K. Momeni, “Effect of the Substrate on MoS2 Monolayer Morphology: An Integrated Computational and Experimental Study.,” ACS applied materials & interfaces. 2022. link Times cited: 6 Abstract: Synthesis of two-dimensional materials, specifically transit… read moreAbstract: Synthesis of two-dimensional materials, specifically transition metal dichalcogenides (TMDs), with controlled lattice orientations is a major barrier to their industrial applications. Controlling the orientation of as-grown TMDs is critical for preventing the formation of grain boundaries, thus reaching their maximum mechanical and optoelectronic performance. Here, we investigated the role of the substrate's crystallinity in the growth orientation of 2D materials using reactive molecular dynamics (MD) simulations and verified with experimental growth using the chemical vapor deposition (CVD) technique. We considered MoS2 as our model material and investigated its growth on crystalline and amorphous silica and sapphire substrates. We revealed the role of the substrate's energy landscape on the orientation of as-grown TMDs, where the presence of monolayer-substrate energy barriers perpendicular to the streamlines hinder the detachment of precursor nuclei from the substrate. We show that MoS2 monolayers with controlled orientations could not be grown on the SiO2 substrate and revealed that amorphization of the substrate changes the intensity and equilibrium distance of monolayer-substrate interactions. Our simulations indicate that 0° rotated MoS2 is the most favorable configuration on a sapphire substrate, consistent with our experimental results. The experimentally validated computational results and insight presented in this study pave the way for the high-quality synthesis of TMDs for high-performance electronic and optoelectronic devices. read less USED (low confidence) H. Ghasemi, S. M. Hatam-Lee, H. K. Tirkolaei, and H. Yazdani, “Biocementation of soils of different surface chemistries via enzyme induced carbonate precipitation (EICP): An integrated laboratory and molecular dynamics study.,” Biophysical chemistry. 2022. link Times cited: 3 USED (low confidence) C. Y. Zhao, Y. Tao, and W. Y. Wang, “Shell effect on microstructure and diffusion in interface region of nanoencapsulated phase change material: a molecular dynamics simulation,” Journal of Molecular Liquids. 2022. link Times cited: 3 USED (low confidence) S. Zhou and H. Shen, “Experimental and numerical studies on micro-bumps without melting of gold films with different thicknesses induced by ultrafast laser,” Optics Communications. 2022. link Times cited: 2 USED (low confidence) H. Zhang, M. Shukla, S. Larson, A. Rajendran, and S. Jiang, “Molecular dynamics study of anisotropic shock responses in oriented α-quartz single crystal,” Journal of Materials Science. 2022. link Times cited: 1 USED (low confidence) Y. Lin and C.-Y. Wu, “Amorphous silica glass nano-grooving behavior investigated using molecular dynamics method,” Proceedings of the Institution of Mechanical Engineers, Part B: Journal of Engineering Manufacture. 2022. link Times cited: 3 Abstract: This work uses molecular dynamics to simulate the grooving b… read moreAbstract: This work uses molecular dynamics to simulate the grooving behavior of optical silica glass. The amorphous SiO2 (silica glass) was fabricated using a melting-quenching process, and the critical cut depth, and mechanism of the chip formation, were explored using a molecular dynamics simulation. The analytical results indicated that the tool edge radius affected the critical cut depth and roughness of the machined surface. A larger tool edge radius had a larger equivalent negative rake angle at the same depth of cut, causing a larger critical cut depth, while producing a smoother machined surface. In addition, the uncut chip thickness affected the tangential force and thrust force distribution weighting of the tool. The temperature field analysis revealed that a groove formed by the chip formation mechanism resulted in a higher workpiece temperature, causing the brittle material to exhibit ductile cutting behavior during the nanogrooving process. However, grooves formed by the scratching-indenting mechanism had a lower workpiece temperature. read less USED (low confidence) M. L. Nietiadi, Y. Rosandi, E. Bringa, and H. Urbassek, “Peripheral Collisions of Ice-covered Silica Dust Grains,” The Astrophysical Journal. 2022. link Times cited: 6 Abstract: Collisions with ice-covered silica grains are studied using … read moreAbstract: Collisions with ice-covered silica grains are studied using molecular-dynamics simulation, with a focus on the influence of the impact parameter on the collision dynamics. The ice mantle induces an attractive interaction between the colliding grains, which is caused by the melting of the mantles in the collision zone and their fusion. For noncentral collisions, this attractive interaction leads to a deflection of the grain trajectories and, at smaller velocities, to the agglomeration (“sticking”) of the colliding grains. The bouncing velocity, which is defined as the smallest velocity at which grains bounce off each other rather than stick, shows only a negligible dependence on the impact parameter. Close to the bouncing velocity, a temporary bridge builds up between the colliding grains, which, however, ruptures when the collided grains separate and relaxes to the grains. At higher velocities, the ice in the collision zone is squeezed out from between the silica cores, forming an expanding disk, which ultimately tears and dissolves into a multitude of small droplets. An essential fraction of the ice cover in the collision zone is then set free to space. Astrophysical implications include the possibility that organic species that might be present in small concentrations on the ice surface or at the ice–silica interface are liberated to space in such noncentral collisions. read less USED (low confidence) S. M. Hatam-Lee, F. Jabbari, and A. Rajabpour, “Interfacial thermal conductance between gold and SiO2: A molecular dynamics study,” Nanoscale and Microscale Thermophysical Engineering. 2022. link Times cited: 2 Abstract: ABSTRACT Silica coating on a gold nanoparticle can improve i… read moreAbstract: ABSTRACT Silica coating on a gold nanoparticle can improve its thermal application in cancer thermotherapy. In this paper, the interfacial thermal conductance between gold and silica is calculated utilizing classical non-equilibrium molecular dynamics. It is revealed that the results of molecular dynamics are different from what has been predicted by the conventional diffuse mismatch model. Furthermore, the interfacial thermal conductance between amorphous SiO2 and gold is approximately twice that of crystalline silica, which is explained by calculating the vibrational density of state mismatches. The interfacial thermal conductance variations in terms of van der Waals interaction strength between gold and silica are also investigated. It is revealed that the conductance increases by about 30% by increasing the simulation temperature from 300 to 700 K. The results of this paper can be useful in nanofluid systems, in addition to the application of silica-coated gold nanoparticles in cancer thermal therapy. read less USED (low confidence) H. Yan, X. Xu, P. Li, P. He, Q. Peng, and C. Ding, “Aluminum Doping Effect on Surface Structure of Silver Ultrathin Films,” Materials. 2022. link Times cited: 1 Abstract: Ultrathin silver films with low loss in the visible and near… read moreAbstract: Ultrathin silver films with low loss in the visible and near-infrared spectrum range have been widely used in the fields of metamaterials and optoelectronics. In this study, Al-doped silver films were prepared by the magnetron sputtering method and were characterized by surface morphology, electrical conductivity, and light transmittance analyses. Molecular dynamics simulations and first-principles density functional theory calculations were applied to study the surface morphologies and migration pathway for the formation mechanisms in Al-doped silver films. The results indicate that the migration barrier of silver on a pristine silver surface is commonly lower than that of an Al-doped surface, revealing that the aluminum atoms in the doping site decrease the surface mobility and are conducive to the formation of small islands of silver. When the islands are dense, they coalesce into a single layer, leading to a smoother surface. This might be the reason for the observably lower 3D growth mode of silver on an Al-doped silver surface. Our results with electronic structure insights on the mechanism of the Al dopants on surface morphologies might benefit the quality control of the silver thin films. read less USED (low confidence) J. Luo, C. Zhou, Q. Li, Y. Cheng, and L. Liu, “Diffusion coefficients of carbon, oxygen and nitrogen in silicon melt,” Journal of Crystal Growth. 2021. link Times cited: 3 USED (low confidence) E. Okulich, V. Okulich, D. Tetelbaum, and A. Mikhaylov, “Simulation of initial stage of silicon cluster formation during post-annealing of memristive structures based on silicon oxide films subjected to Si+ implantation,” Materials Letters. 2021. link Times cited: 1 USED (low confidence) Z. Wu and L. Zhang, “Mechanical properties and deformation mechanisms of surface-modified 6H-silicon carbide,” Journal of Materials Science & Technology. 2021. link Times cited: 15 USED (low confidence) D. Bayer-Buhr, M. Vimal, A. Prakash, U. Gross, and T. Fieback, “Determination of thermal accommodation coefficients on CaSiO3 and SiO2 using molecular dynamics and experiments,” International Journal of Heat and Mass Transfer. 2021. link Times cited: 2 USED (low confidence) Z. Ma, R. Gamage, and C. Zhang, “Mechanical properties of α-quartz using nanoindentation tests and molecular dynamics simulations,” International Journal of Rock Mechanics and Mining Sciences. 2021. link Times cited: 13 USED (low confidence) A. Lahti, R. Östermark, and K. Kokko, “Optimization of SiO2 with GHA and basin hopping,” Computational Materials Science. 2021. link Times cited: 1 USED (low confidence) F. Molaei, “Molecular dynamics simulation of edge crack propagation in single crystalline alpha quartz.,” Journal of molecular graphics & modelling. 2021. link Times cited: 6 USED (low confidence) M. Y. Yang, Q. Sheng, H. Zhang, and G. Tang, “Water molecular bridge undermines thermal insulation of Nano-porous silica aerogels,” Journal of Molecular Liquids. 2021. link Times cited: 12 USED (low confidence) P. N. Wimalasiri, N. P. Nguyen, H. S. Senanayake, B. Laird, and W. Thompson, “Amorphous Silica Slab Models with Variable Surface Roughness and Silanol Density for Use in Simulations of Dynamics and Catalysis,” The Journal of Physical Chemistry C. 2021. link Times cited: 9 USED (low confidence) L. Qiufa, J. Lu, Z. Tian, and F. Jiang, “Controllable material removal behavior of 6H-SiC wafer in nanoscale polishing,” Applied Surface Science. 2021. link Times cited: 19 USED (low confidence) Z. Li, H. Wang, H. Zhao, J. Wang, X. Wei, and H. Gu, “Effect of regulating compressive strains on thermal transport of silicon-based amorphous silica thin films and interfacial thermal resistance,” Vacuum. 2021. link Times cited: 0 USED (low confidence) H. Wang, J. Zhao, B. Wang, H. Qi, and J. Shao, “Structure and shock properties of amorphous silica predicted by a metal-organic framework force field,” Optical Materials Express. 2021. link Times cited: 0 Abstract: The laser damage induced by nano-absorbing centers generally… read moreAbstract: The laser damage induced by nano-absorbing centers generally results in a local high temperature and pressure environment, leading to denser phases and complex hydrodynamic processes. Here we parameterize the metal-organic framework force field to overcome the notorious unphysical agglomeration at small atomic distance in a Buckingham term. The structure and shock properties of amorphous silica are predicted well by the parameterized force field. By avoiding the Ewald summation of long-range coulomb interaction, the periodic boundary condition is not in such demand that the computational efficiency is greatly improved. The parameterized force field implicates a prospect for the atomic investigation of laser-induced hydrodynamic processes around the free surface or interface. read less USED (low confidence) S. Uchida, K. Fujiwara, and M. Shibahara, “Structure of the Water Molecule Layer between Ice and Amorphous/Crystalline Surfaces Based on Molecular Dynamics Simulations.,” The journal of physical chemistry. B. 2021. link Times cited: 2 Abstract: The structure of the water layer between the ice interface a… read moreAbstract: The structure of the water layer between the ice interface and the hydroxylated amorphous/crystalline silica surfaces was investigated using molecular dynamics simulations. The results indicate that the density profile in the direction perpendicular to the surface has two density peaks in the water layer at the ice-silica interface, which are affected by the silanol group density on the wall and the degree of supercooling in the system. In the two density peaks, the one facing the ice interface side has the same structure as the ice crystal, while the other density peak facing the silica surface has an icelike structure. In the solidification process, the ice and icelike structures in the layer progress more on the amorphous silica surface where the density of the silanol groups is low. The relationship between the ice crystallization and the thickness of the layer has been studied in detail; the lower the temperature, the more the ice crystallization progresses and the thinner the layer becomes. read less USED (low confidence) A. Kharin, M. Grigoryeva, I. Zavestovskaya, and V. Timoshenko, “Effect of silicon target porosity on laser ablation threshold: molecular dynamics simulation,” Laser Physics Letters. 2021. link Times cited: 1 Abstract: Ablation of a porous silicon target under irradiation with u… read moreAbstract: Ablation of a porous silicon target under irradiation with ultrashort laser pulse is simulated by means of the molecular dynamics approach. The number of ablated atoms is calculated for targets with different porosity under irradiation with wavelengths in ultraviolet (UV) and visible spectral ranges, which correspond to stronger and weaker absorption coefficient, respectively. An increase of the porosity to 80% leads to 1.5–3 times decrease of the ablation threshold compared to the bulk silicon, while a decrease of pores size from 2.5 to 1.2 nm leads to the stronger ablation threshold drop and the effect is stronger for the UV irradiation. The results are useful for laser processing of silicon-based targets and nanofabrication. read less USED (low confidence) H. Vázquez and F. Djurabekova, “Ultrafast phase transitions in polyamorphic materials triggered by swift heavy ion impacts,” Physical Review Materials. 2021. link Times cited: 1 USED (low confidence) S. Fujii and A. Seko, “Structure and lattice thermal conductivity of grain boundaries in silicon by using machine learning potential and molecular dynamics,” Computational Materials Science. 2021. link Times cited: 8 USED (low confidence) J. Chen, L. Fang, M. Zhang, W. Peng, K. Sun, and J. Han, “Stress Relaxation Behaviors of Monocrystalline Silicon Coated with Amorphous SiO$_2$ Film: A Molecular Dynamics Study,” Acta Mechanica Solida Sinica. 2021. link Times cited: 3 USED (low confidence) Y.-K. Weng, A. Y. Nobakht, S. Shin, K. Kihm, and D. Aaron, “Effects of mass and interaction mismatches on in-plane and cross-plane thermal transport of Si-doped graphene,” International Journal of Heat and Mass Transfer. 2021. link Times cited: 12 USED (low confidence) J. Martínez-González, N. J. English, and A. Gowen, “Molecular simulation of water adsorption on hydrophilic and hydrophobic surfaces of silicon: IR-spectral explorations,” Molecular Simulation. 2021. link Times cited: 2 Abstract: ABSTRACT Molecular-dynamics simulations have been performed … read moreAbstract: ABSTRACT Molecular-dynamics simulations have been performed for full liquid water adsorbed onto two planar silicon surfaces, with varying hydrogen- and hydroxyl-termination (mimicking different extents of hydrophobicity and hydrophophilicity). It was found that there was water-density ‘ordering’ perpendicular to both surfaces – heavily dependent on the degree of hydrophobicity. The position and the width of the three solvation layers closest to the different surfaces depends, again, on the hydrophobicity of the surface. IR spectra of the first monolayer of adsorbed water indicate similarities to more confined-water dynamical behaviour, but without becoming ice-like. Moving away from the surface, the water behaviour converges on that of liquid water, albeit with some intermediate characteristics; this was seen for both hydro-phobic and –philic surfaces. read less USED (low confidence) M. Eghbalian, R. Ansari, and S. Rouhi, “Mechanical properties of oxygen-functionalized silicon carbide nanotubes: A molecular dynamics study,” Physica B-condensed Matter. 2021. link Times cited: 11 USED (low confidence) Y. Yu, “Deposited Mono-component Cu Metallic Glass: A Molecular Dynamics Study,” Materials today communications. 2021. link Times cited: 4 USED (low confidence) Z. Yin, Y. Yu, H.-C. Chen, J. Li, and L. Bai, “Nanofriction behaviors between silicon-doped diamond-like carbon films under different testing conditions,” Computational Materials Science. 2021. link Times cited: 7 USED (low confidence) M. L. Nietiadi, Y. Rosandi, and H. Urbassek, “Collisions between ice-covered silica grains: An atomistic study,” Icarus. 2020. link Times cited: 7 USED (low confidence) W. Sun, J. Jiang, and P. Chen, “Microstructural evolution, shocking sintering mechanism and dynamic mechanical behaviours of silica nanoparticles acting as catalyst carrier in energetic nanomaterials during shock-wave impact.” 2020. link Times cited: 1 USED (low confidence) Z. Ma, R. Gamage, and C. Zhang, “Effects of temperature and grain size on the mechanical properties of polycrystalline quartz,” Computational Materials Science. 2020. link Times cited: 16 USED (low confidence) H. Gu, J. Wang, X. Wei, H. Wang, and Z. Li, “Thermal conductivity and interfacial thermal resistance in the heterostructure of silicon/amorphous silicon dioxide: the strain and temperature effect,” Nanotechnology. 2020. link Times cited: 10 Abstract: This article reports the thermal conduction properties of Si… read moreAbstract: This article reports the thermal conduction properties of Si/a-SiO2 heterostructure with two different interfaces: weak and strong coupling strength through molecular dynamics simulation. The size and temperature dependencies on the interfacial thermal resistance of the weak coupling interface are larger than those of the strong coupling interface. The thermal conduction in Si/a-SiO2 shows strong anisotropy. The thermal conductivity, interfacial thermal resistance, and enhancement of the anisotropy can be modulated by changing the strains applied to the heterostructures. This work provides an optional way to design the silicon-based heterostructures considering heat insulation and heat dissipation. read less USED (low confidence) I. Bejenari, A. Burenkov, P. Pichler, I. Deretzis, and A. L. Magna, “Molecular Dynamics Modeling of the Radial Heat Transfer from Silicon Nanowires,” 2020 International Conference on Simulation of Semiconductor Processes and Devices (SISPAD). 2020. link Times cited: 2 Abstract: Thermal transport in radial direction in Si nanowires embedd… read moreAbstract: Thermal transport in radial direction in Si nanowires embedded into amorphous silicon dioxide has been studied using nonequilibrium molecular dynamics simulations. For comparison, we also considered the axial heat transfer. For Si nanowires with a radius of 2.6 nm, both radial and axial thermal conductivities were found to be about independent of the SiO2 thickness ranging from 1 nm to 3 nm. The radial thermal conductivity of the Si core and of the covering SiO2 material are similar and nearly equal to 1 $W\cdot K^{-1}\cdot m^{-1}$. Thermal resistances for the heat transfer from uniformly heated nanowires in radial direction are by a factor of 3 to 4 lower than those for the heat transfer in axial direction. read less USED (low confidence) X. Cui, X. Cheng, H.-hua Xu, B. Li, and J. Zhu, “Enhancement of thermophysical coefficients in nanofluids: A simulation study,” International Journal of Modern Physics B. 2020. link Times cited: 4 Abstract: Molten salts constitute one kind of PCMs (Phase Change Mater… read moreAbstract: Molten salts constitute one kind of PCMs (Phase Change Materials) widely used in concentrating solar power facilities for heat storage and heat transfer. This paper aims to simulate nanofluid PCMs ... read less USED (low confidence) Y. Jiao and J. Fish, “Coupled thermodynamically consistent thermo-mechanical model of silica glass subjected to hypervelocity impact,” Computer Methods in Applied Mechanics and Engineering. 2020. link Times cited: 3 USED (low confidence) C. Liu, J. Chu, X. Chen, J. Xiao, and J. Xu, “Molecular dynamics simulation on structure evolution of silica glass in nano-cutting at high temperature,” Molecular Simulation. 2020. link Times cited: 7 Abstract: ABSTRACT Machining of silica glass is indeed a challenge due… read moreAbstract: ABSTRACT Machining of silica glass is indeed a challenge due to its extreme brittleness and hardness. In recent years, laser-assisted machining (LAM) is becoming a powerful method for cutting silica glass. However, as a typical non-crystalline structure, the deformation mechanism of silica glass in the cutting process at high temperature has not been explored clearly. In this paper, classical molecular dynamics simulation was conducted to investigate the structure evolution of silica glass during the nano-cutting process at high temperature. Firstly, a uniaxial tension test was carried to investigate the brittle-to-ductile transition with increasing temperature. Then, the cutting simulation was conducted at 300 and 1500 K. The results showed that the plastic deformation is promoted at 1500 K. The atomic flow of silica glass is different from crystal material during the cutting process. No rotational flow and upward motion of atoms are observed. Moreover, microstructure evolution like bond-switch and rings distribution was discussed in detail. Furthermore, although the densification scope is almost unaffected, the extent of densification near the machined surface is greatly decreased at a high temperature. These results contribute to the theoretical research on the precision machining of silica glass with LAM. read less USED (low confidence) R. Shen, Q. Bai, Y. Li, Y.-bo Guo, and F.-hu Zhang, “Ejecta distribution and transport property of fused silica under the laser shock loading,” Journal of Applied Physics. 2020. link Times cited: 3 Abstract: Laser-induced particle ejection on the exit surface of fused… read moreAbstract: Laser-induced particle ejection on the exit surface of fused silica serves as an important contaminant source in a high-power laser system. The transport process of molten silica particles in a gas environment or vacuum is important in understanding the change in size and temperature of silica particles, which influence the ultra-clean manufacturing of optical components. In this paper, the ejection process of fused silica is investigated using molecular dynamics simulation. The results show that the geometry of a surface scratch influences the mass of the microjet. With shallower groove depth and a smaller vortex angle, the mass of the microjet is less under shock loading. The size of ejected particles tends to decrease gradually and does not change any more eventually. Besides, these particles become dispersed during the transport process in a vacuum. On the other hand, background gas suppresses the particle flow and slows down the particle flow. As the ejected particles compress gas, vapor and small clusters (N < 50) are stripped from the microjet continuously. Eventually, the number of nanoparticles that exceed the free surface decreases to zero. The stripped small clusters behind the head of the microjet recombine with other clusters, which change the volume density of ejected particles near the free surface. The higher velocity of ejected particles induces a stronger gas stripping effect, which makes an increase in the number of small clusters (N < 50). The results can help understand the behavior of particle ejection and the transport process of silica particles in a gas environment or vacuum, especially in the field of laser-induced particle ejection on the exit surface or the laser ablation of fused silica producing aerosol. read less USED (low confidence) A. Sycheva and E. Voronina, “Features of Low-Energy He and Ar Ion Irradiation of Nanoporous Si/SiO2-Based Materials,” Technical Physics Letters. 2020. link Times cited: 0 USED (low confidence) W. Zhao and F. Duan, “Friction properties of carbon nanoparticles (nanodiamond and nanoscroll) confined between DLC and a-SiO2 surfaces,” Tribology International. 2020. link Times cited: 21 USED (low confidence) J. Chen, L. Fang, K. Sun, and J. Han, “Creep behaviors of surface-modified silicon: A molecular dynamics study,” Computational Materials Science. 2020. link Times cited: 6 USED (low confidence) Y. Zheng et al., “Disordered hyperuniformity in two-dimensional amorphous silica,” Science Advances. 2020. link Times cited: 32 Abstract: Two-dimensional amorphous silica exhibits a novel state of m… read moreAbstract: Two-dimensional amorphous silica exhibits a novel state of matter, disordered hyperuniformity, and enhanced electronic transport. Disordered hyperuniformity (DHU) is a recently proposed new state of matter, which has been observed in a variety of classical and quantum many-body systems. DHU systems are characterized by vanishing infinite-wavelength normalized density fluctuations and are endowed with unique novel physical properties. Here, we report the discovery of disordered hyperuniformity in atomic-scale two-dimensional materials, i.e., amorphous silica composed of a single layer of atoms, based on spectral-density analysis of high-resolution transmission electron microscopy images. Moreover, we show via large-scale density functional theory calculations that DHU leads to almost complete closure of the electronic bandgap compared to the crystalline counterpart, making the material effectively a metal. This is in contrast to the conventional wisdom that disorder generally diminishes electronic transport and is due to the unique electron wave localization induced by the topological defects in the DHU state. read less USED (low confidence) M. L. Nietiadi, Y. Rosandi, and H. Urbassek, “Bouncing of Hydroxylated Silica Nanoparticles: an Atomistic Study Based on REAX Potentials,” Nanoscale Research Letters. 2020. link Times cited: 10 USED (low confidence) W. Sun, J. Jiang, and P. Chen, “Dynamic mechanical contact behaviours of amorphous nanoparticles subjected to high-speed impact,” Powder Technology. 2020. link Times cited: 6 USED (low confidence) E. Voronina et al., “Pore sealing mechanism in OSG low‐
k
films under ion bombardment,” Plasma Processes and Polymers. 2020. link Times cited: 5 USED (low confidence) P. Valerius et al., “Reversible crystalline-to-amorphous phase transformation in monolayer MoS2 under grazing ion irradiation,” 2D Materials. 2020. link Times cited: 13 Abstract: By combining scanning tunneling microscopy, low-energy elect… read moreAbstract: By combining scanning tunneling microscopy, low-energy electron diffraction, photoluminescence and Raman spectroscopy experiments with molecular dynamics simulations, a comprehensive picture of the structural and electronic response of a monolayer of MoS2 to 500 eV Xe+ irradiation is obtained. The MoS2 layer is epitaxially grown on graphene/Ir(1 1 1) and analyzed before and after irradiation in situ under ultra-high vacuum conditions. Through optimized irradiation conditions using low-energy ions with grazing trajectories, amorphization of the monolayer is induced already at low ion fluences of ions cm−2 and without inducing damage underneath the MoS2 layer. The crystalline-to-amorphous transformation is accompanied by changes in the electronic properties from semiconductor-to-metal and an extinction of photoluminescence. Upon thermal annealing, the re-crystallization occurs with restoration of the semiconducting properties, but residual defects prevent the recovery of photoluminescence. read less USED (low confidence) F. Djurabekova, C. Fridlund, and K. Nordlund, “Defect and density evolution under high-fluence ion irradiation of
Si/SiO2
heterostructures,” Physical Review Materials. 2020. link Times cited: 5 Abstract: We present molecular dynamics simulations of atomic mixing o… read moreAbstract: We present molecular dynamics simulations of atomic mixing over a Si/SiO2 heterostructure interface, induced by focused Ne+ and broad Si+ ion-beam irradiations, using a speed-up scheme that significantly reduces the relaxation time of the cascading recoils. To assess the qualitative reliance of the chosen method, two different potential models for Si–O, Si–Si, and O–O interactions were used: the Stillinger-Weber–like Watanabe-Samela potential and the Tersoff-like Munetoh potential. Furthermore, the molecular dynamics simulations were assessed by simulating a similar case, at a total fluence of 1×1015 cm−2, with the binary collision approximation. The same general atomic density profile distributions were achieved with both models; however, the binary collision approach showed shallower penetration of Si into the SiO2 layer. Coordination analysis of the molecular dynamics results provides strong evidence that ion mixing at high fluences leads to coordination defects, which will affect the electronic properties of the structures unless removed with annealing. read less USED (low confidence) T. Vo, B. Reeder, A. Damone, and P. Newell, “Effect of Domain Size, Boundary, and Loading Conditions on Mechanical Properties of Amorphous Silica: A Reactive Molecular Dynamics Study,” Nanomaterials. 2019. link Times cited: 11 Abstract: Mechanical properties are very important when choosing a mat… read moreAbstract: Mechanical properties are very important when choosing a material for a specific application. They help to determine the range of usefulness of a material, establish the service life, and classify and identify materials. The size effect on mechanical properties has been well established numerically and experimentally. However, the role of the size effect combined with boundary and loading conditions on mechanical properties remains unknown. In this paper, by using molecular dynamics (MD) simulations with the state-of-the-art ReaxFF force field, we study mechanical properties of amorphous silica (e.g., Young’s modulus, Poisson’s ratio) as a function of domain size, full-/semi-periodic boundary condition, and tensile/compressive loading. We found that the domain-size effect on Young’s modulus and Poisson’s ratio is much more significant in semi-periodic domains compared to full-periodic domains. The results, for the first time, revealed the bimodular and anisotropic nature of amorphous silica at the atomic level. We also defined a “safe zone” regarding the domain size, where the bulk properties of amorphous silica can be reproducible, while the computational cost and accuracy are in balance. read less USED (low confidence) N. Liao, B. Zheng, M. Zhang, and W. Xue, “Numerical approach to evaluate performance of porous SiC5/4O3/2 as potential high temperature hydrogen gas sensor,” International Journal of Hydrogen Energy. 2019. link Times cited: 16 USED (low confidence) P. Maximiano, L. Durães, and P. Simões, “Overview of Multiscale Molecular Modeling and Simulation of Silica Aerogels,” Industrial & Engineering Chemistry Research. 2019. link Times cited: 11 Abstract: Molecular simulation has become an integral and invaluable p… read moreAbstract: Molecular simulation has become an integral and invaluable part of Chemical Product Engineering, as it provides fundamental and indispensable insights for a rational product design. Silica aerogels... read less USED (low confidence) Y. Wan, Y. Gao, J. Wang, Y.-qing Yang, and Z. Xia, “Rapid Water Harvesting and Non-thermal Drying in Humid Air by N-doped Graphene Micro-Pads.,” Langmuir : the ACS journal of surfaces and colloids. 2019. link Times cited: 6 Abstract: We demonstrated a novel nano-textured graphene micro-pad tha… read moreAbstract: We demonstrated a novel nano-textured graphene micro-pad that can rapidly harvest water from the air to generate microscale water droplets with the desired size in designated positions on demand by simply applying a negative electric bias of -1.5 ~ -15V. More interestingly, the water droplets can be reversibly dried non-thermally with the pad at ambient temperature in humid air (~ 85%RH) by applying positive electric bias of +1.5 ~ +15V. The harvesting and drying rates on the glass are 2.7 m3/s and 1.5 m3/s under a bias of -15V and +15V, respectively, but no apparent harvesting or drying activities were observed without the bias. The energy consumption is minimal as there is no Joule current due to the insulative substrate. It was shown that substrate wettability and ions play an important role enabling the fast water harvesting and non-thermal drying. Molecular modeling is developed to understand the harvesting and drying mechanisms at the atomic scale. The water harvesting/drying approach may be useful for many technological applications such as micro-/nanolithography, 3D printing, MEMS, biochemical and microfluid devices. read less USED (low confidence) Z. Liu, C. Ji, B. Wang, and S. Sun, “Role of a nanoparticle on ultrasonic cavitation in nanofluids,” Micro & Nano Letters. 2019. link Times cited: 5 Abstract: Ultrasonic cavitation in nanofluids improves material remova… read moreAbstract: Ultrasonic cavitation in nanofluids improves material removal rate and surface quality. Ultrasonic cavitation in nanofluids was investigated using molecular dynamics simulations. The formation and growth of nanobubbles were promoted, by nanoparticles in water systems. Three distinct impact stages were observed which were caused by the impact of the shock wave, nanojet and nanoparticle. These differed in the system without a nanoparticle. The material removal rate was primarily caused by the nanoparticle hit, a result of the nanobubble collapsing during the third impact. A mechanism of material removal in ultrasonic polishing is discussed at a nanometric level. read less USED (low confidence) N. Hong, L. T. Vinh, P. K. Hung, M. Dung, and N. V. Yên, “The structural transition under densification and the relationship between structure and density of silica glass,” The European Physical Journal B. 2019. link Times cited: 4 USED (low confidence) C. Ji, S. Sun, B. Wang, and B. Lin, “Molecular dynamic simulations of the roles of nanoparticles in sliding friction process,” Chemical Physics Letters. 2019. link Times cited: 9 USED (low confidence) P. K. Hung, L. T. Vinh, N. Hong, G. Trang, and N. T. Nhan, “Insight into microstructure and dynamics of network forming liquid from the analysis based on shell–core particles,” The European Physical Journal B. 2019. link Times cited: 3 USED (low confidence) J. Morthomas, W. Gonçalves, M. Perez, G. Foray, C. L. Martin, and P. Chantrenne, “A novel method to predict the thermal conductivity of nanoporous materials from atomistic simulations,” Journal of Non-Crystalline Solids. 2019. link Times cited: 12 USED (low confidence) I. Ponomarev, A. V. van Duin, and P. Kroll, “Reactive Force Field for Simulations of the Pyrolysis of Polysiloxanes into Silicon Oxycarbide Ceramics,” The Journal of Physical Chemistry C. 2019. link Times cited: 18 Abstract: We provide a new reactive force field (ReaxFF) for simulatio… read moreAbstract: We provide a new reactive force field (ReaxFF) for simulations of silicon oxycarbide (SiCO) ceramics and of their syntheses from inorganic polymer precursors. The validity of the force field is extensively tested against experimental and computational thermochemical data. Its performance in simulation at elevated temperatures is gauged by the results of comprehensive ab initio molecular dynamics simulations. We apply the force field to the formation of amorphous SiCO in a simulated polymer pyrolysis. Modeling results are in good agreement with experimental observations and allow new insights into the formation of graphene segregations embedded in an amorphous oxycarbide matrix. The new ReaxFF for Si–C–O–H compounds enables large-scale and long-time atomistic simulations with unprecedented fidelity. read less USED (low confidence) J. Moon, R. Hermann, M. Manley, A. Alatas, A. Said, and A. Minnich, “Thermal acoustic excitations with atomic-scale wavelengths in amorphous silicon,” Physical Review Materials. 2019. link Times cited: 18 Abstract: The vibrational properties of glasses remain a topic of inte… read moreAbstract: The vibrational properties of glasses remain a topic of intense interest due to several unresolved puzzles, including the origin of the Boson peak and the mechanisms of thermal transport. Inelastic scattering measurements have revealed that amorphous solids support collective acoustic excitations with low THz frequencies despite the atomic disorder, but these frequencies are well below most of the thermal vibrational spectrum. Here, we report the observation of acoustic excitations with frequencies up to 10 THz in amorphous silicon. The excitations have atomic-scale wavelengths as short as 6 A and exist well into the thermal vibrational frequencies. Simulations indicate that these high-frequency waves are supported due to the high group velocity and monatomic composition of a-Si, suggesting that other glasses with these characteristics may also exhibit such excitations. Our findings demonstrate that a substantial portion of thermal vibrational modes in amorphous materials can still be described as a phonon gas despite the lack of atomic order. read less USED (low confidence) S. Gelin, D. Poinot, S. Châtel, P. Calba, and A. Lemaître, “Microstructural origin of compressive
in situ
stresses in electron-gun-evaporated silica thin films,” Physical Review Materials. 2019. link Times cited: 1 USED (low confidence) P. K. Hung, N. Hong, G. Trang, and T. Iitaka, “Topological analysis on structure and dynamics of SiO2 liquid with the help of Si-particle and O-particle statistics,” Materials Research Express. 2019. link Times cited: 2 Abstract: A large model of silica liquid has been produced at temperat… read moreAbstract: A large model of silica liquid has been produced at temperature of 3000 K and ambient pressure by molecular dynamics (MD) simulation. We propose a topological analysis based on shell-core particles. Our result shows that the dynamics is spatially sparse due to the SiO4 ↔ SiO5 and OSi2 ↔ OSi3 transformations happen non-uniformly in the network structure. The simulation reveals Si-particles having 2 Si and O-particles containing up to 14 O. The network structure comprises clusters of particles which occupy micro-regions with pure compositions. The number of atoms in a Si-cluster varies from 2 to 7, and the O-cluster contains up to 63 atoms. We also found particles and clusters of particles which are stable for a long time. The stable particle and cluster of particles occupy micro-regions where the atoms of the same ion type are confined. The strong chemical Si–O bonds between core and shell atoms prevent those particles from breaking apart. The atoms of rigid SiO4 tetrahedrons are not uniformly distributed in the network structure, but instead they gather into domains containing the rigid Si–O subnets, stable particles and clusters of stable particles. read less USED (low confidence) A. Sycheva, E. Voronina, T. Rakhimova, and A. Rakhimov, “Influence of porosity and pore size on sputtering of nanoporous structures by low-energy Ar ions: Molecular dynamics study,” Applied Surface Science. 2019. link Times cited: 13 USED (low confidence) Y. Wan, Y. Gao, and Z. Xia, “Highly Switchable Adhesion of N-Doped Graphene Interfaces for Robust Micromanipulation.,” ACS applied materials & interfaces. 2019. link Times cited: 7 Abstract: We demonstrated an N-doped graphene interface with highly sw… read moreAbstract: We demonstrated an N-doped graphene interface with highly switchable adhesion and robust micromanipulation capability triggered by external electric signals. Upon applying a small dc or ac electrical bias, this nanotextured surface can collect environmental moisture to form a large number of water bridges between the graphene and target surface, which lead to a drastic change in adhesive force. Turning on and off the electrical bias can control this graphene interface as a robust micro/nanomanipulator to pick up and drop off various micro/nano-objects for precise assembling. Molecular dynamics simulation reveals that the electrically induced electric double layer and ordered icelike structures at the graphene-water interface strengthen the water bridges and consequently enhance force switchability. In addition to the micro-/nanomanipulation, this switchable adhesion may have many technical implications such as climbing robots, sensors, microfluidic devices, and advanced drug delivery. read less USED (low confidence) M. Dholakia, S. Chandra, and S. M. Jaya, “A comparative study of topology and local disorder in Y 2 O 3, Y 2 TiO 5, and Y 2 Ti 2 O 7 crystals,” Journal of Applied Physics. 2019. link Times cited: 1 Abstract: Y 2 O 3, Y 2 TiO 5, and Y 2 Ti 2 O 7 precipitates are the ke… read moreAbstract: Y 2 O 3, Y 2 TiO 5, and Y 2 Ti 2 O 7 precipitates are the key ingredients of the oxide dispersion strengthened (ODS) steels, and a study of their irradiation behavior and amorphization resistance is very important for the nuclear industry. We perform molecular dynamics simulations of disorder induced by displacement cascades and by equilibrating the systems at high temperatures and study the topology and defect morphology using various analyses. The studies show that a radiation damaged and a melted system can have very different topologies, and this difference can be effectively discerned by a topology based analysis. It is shown that all the three systems are constrained and quite resistant to amorphization; however, Y 2 O 3 and Y 2 Ti 2 O 7 have a better resistance to amorphization as compared to Y 2 TiO 5. Y 2 O 3, Y 2 TiO 5, and Y 2 Ti 2 O 7 precipitates are the key ingredients of the oxide dispersion strengthened (ODS) steels, and a study of their irradiation behavior and amorphization resistance is very important for the nuclear industry. We perform molecular dynamics simulations of disorder induced by displacement cascades and by equilibrating the systems at high temperatures and study the topology and defect morphology using various analyses. The studies show that a radiation damaged and a melted system can have very different topologies, and this difference can be effectively discerned by a topology based analysis. It is shown that all the three systems are constrained and quite resistant to amorphization; however, Y 2 O 3 and Y 2 Ti 2 O 7 have a better resistance to amorphization as compared to Y 2 TiO 5. read less USED (low confidence) N. A. Mehta, A. Rayabharam, and D. Levin, “Correction: Simulations of surface evolution due to particulate-surface interaction,” AIAA Scitech 2019 Forum. 2019. link Times cited: 0 USED (low confidence) J. Chen et al., “Effect of indentation speed on deformation behaviors of surface modified silicon: A molecular dynamics study,” Computational Materials Science. 2018. link Times cited: 16 USED (low confidence) A. Antidormi et al., “Physical and Chemical Control of Interface Stability in Porous Si–Eumelanin Hybrids,” The Journal of Physical Chemistry C. 2018. link Times cited: 12 Abstract: The organic/inorganic interface in thin nanosized porous str… read moreAbstract: The organic/inorganic interface in thin nanosized porous structures has a key role in determining the final properties of the composite materials. By use of the porous silicon/eumelanin hybrids as a case study, the role of this interface was investigated by experimental and computational methods. Our results show that an increased polymer density close to the hybrid interface strongly modifies the diffusion of the chemical species within the polymer molecule, affecting then the oxidation level of the pores’ inner Si surface. We observed a greater stability induced by increased pore diameter, a behavior that with computational and chemical arguments we attributed to a modified diffusion of the hydrogen peroxide toward the Si/eumelanin interface. Our results show that the overall behavior of a polymer when inserted in a tiny nanoscale structure must be taken into account for a correct understanding and control of the hybrids properties and that the formation of the interface alone may not be sufficient. read less USED (low confidence) Z. Wang et al., “Liquid-liquid phase transition in nanoconfined Si-rich SiO2 liquids,” Computational Materials Science. 2018. link Times cited: 2 USED (low confidence) Y. Zhang, M. Li, Y. Gao, B. Xu, and W. Lu, “Compressing liquid nanofoam systems: liquid infiltration or nanopore deformation?,” Nanoscale. 2018. link Times cited: 13 Abstract: Understanding the invasion of a liquid into porous structure… read moreAbstract: Understanding the invasion of a liquid into porous structures is the foundation of the characterization of the porosity-related properties of materials and is also of fundamental importance in the design of porous solid-liquid enabled energy protection systems, yet whether solid pores deform has been unclear so far. Here, we present a competition mechanism between liquid infiltration and cell wall buckling deformation by investigating a liquid nanofoam (LN) system subjected to quasi-static compression. The critical buckling stress of the cell wall and the infiltration pressure of liquid invasion into nanopores are studied and correlated through numerical simulation and experimental validation to reveal the quantitative relationship between nanopore deformation and liquid invasion. The analysis shows that liquid infiltration occurs, independent of the axial buckling stress of the cell wall; in contrast, the nanopore collapses radially when the radial collapse pressure is lower than the pressure of liquid infiltration, preventing the liquid invasion. Comprehensive molecular dynamics (MD) simulations are performed and demonstrate the deformation behavior of nanopores and cell wall-liquid interactions in a broad range. Pressure-induced compression experiments on a silica-based LN system are carried out and validate these theoretical and MD results. read less USED (low confidence) L.-B. Shi, S. Cao, J. Zhang, X. Xiu, and H. Dong, “Mechanical behaviors and electronic characteristics on two-dimensional C2N3 and C2N3H: First principles calculations,” Physica E: Low-dimensional Systems and Nanostructures. 2018. link Times cited: 12 USED (low confidence) H. Peer-Mohammadi, A. Rajabpour, and M. Khanaki, “Grain size facilitating the heat transfer between graphene and silica substrate,” Computational Materials Science. 2018. link Times cited: 16 USED (low confidence) T. Li, Z. Tang, Z. Huang, and J. Yu, “Substrate effects on the thermal performance of in-plane graphene/hexagonal boron nitride heterostructures,” Carbon. 2018. link Times cited: 27 USED (low confidence) H. Gu and H. Wang, “Effect of strain on thermal conductivity of amorphous silicon dioxide thin films: A molecular dynamics study,” Computational Materials Science. 2018. link Times cited: 15 USED (low confidence) P. Mota‐Santiago et al., “Nanoscale density variations induced by high energy heavy ions in amorphous silicon nitride and silicon dioxide,” Nanotechnology. 2018. link Times cited: 25 Abstract: The cylindrical nanoscale density variations resulting from … read moreAbstract: The cylindrical nanoscale density variations resulting from the interaction of 185 MeV and 2.2 GeV Au ions with 1.0 μm thick amorphous SiNx:H and SiOx:H layers are determined using small angle x-ray scattering measurements. The resulting density profiles resembles an under-dense core surrounded by an over-dense shell with a smooth transition between the two regions, consistent with molecular-dynamics simulations. For amorphous SiNx:H, the density variations show a radius of 4.2 nm with a relative density change three times larger than the value determined for amorphous SiOx:H, with a radius of 5.5 nm. Complementary infrared spectroscopy measurements exhibit a damage cross-section comparable to the core dimensions. The morphology of the density variations results from freezing in the local viscous flow arising from the non-uniform temperature profile in the radial direction of the ion path. The concomitant drop in viscosity mediated by the thermal conductivity appears to be the main driving force rather than the presence of a density anomaly. read less USED (low confidence) P. Ranjan, R. Balasubramaniam, and V. Jain, “Investigations into the mechanism of material removal and surface modification at atomic scale on stainless steel using molecular dynamics simulation,” Philosophical Magazine. 2018. link Times cited: 21 Abstract: A molecular dynamics simulation (MDS) has been carried out t… read moreAbstract: A molecular dynamics simulation (MDS) has been carried out to investigate the material removal phenomenon of chemo-mechanical magnetorheological finishing (CMMRF) process. To understand the role of chemical assisted mechanical abrasion in CMMRF process, material removal phenomenon is subdivided into three different stages. In the first stage, new atomic bonds viz. Fe–O–Si is created on the surface of the workpiece (stainless steel). The second stage deals with the rupture of parent bonds like Fe–Fe on the workpiece. In the final stage, removal of material from the surface in the form of dislodged debris (cluster of atoms) takes place. Effects of process parameters like abrasive particles, depth of penetration and initial surface condition on finishing force, potential energy (towards secondary phenomenon such as chemical instability of the finished surface) and material removal at atomic scale have been investigated. It was observed that the type of abrasive particle is one of the important parameters to produce atomically smooth surface. Experiments were also conducted as per the MDS to generate defect-free and sub-nanometre-level finished surface (Ra value better than 0.2 nm). The experimental results reasonably agree well with the simulation results. read less USED (low confidence) H. Farahani, A. Rajabpour, M. Khanaki, and A. Reyhani, “Interfacial thermal resistance between few-layer MoS2 and silica substrates: A molecular dynamics study,” Computational Materials Science. 2018. link Times cited: 34 USED (low confidence) T. Oyake, L. Feng, T. Shiga, M. Isogawa, Y. Nakamura, and J. Shiomi, “Ultimate Confinement of Phonon Propagation in Silicon Nanocrystalline Structure.,” Physical review letters. 2018. link Times cited: 39 Abstract: Temperature-dependent thermal conductivity of epitaxial sili… read moreAbstract: Temperature-dependent thermal conductivity of epitaxial silicon nanocrystalline (SiNC) structures composed of nanometer-sized grains separated by ultrathin silicon-oxide (SiO_{2}) films (∼0.3 nm) is measured by the time domain thermoreflectance technique in the range from 50 to 300 K. The thermal conductivity of SiNC structures with a grain size of 3 and 5 nm is anomalously low at the entire temperature range, significantly below the values of bulk amorphous Si and SiO_{2}. The phonon gas kinetic model, with intrinsic transport properties obtained by first-principles-based anharmonic lattice dynamics and phonon transmittance across ultrathin SiO_{2} films obtained by atomistic Green's function, reproduces the measured thermal conductivity without any fitting parameters. The analysis reveals that mean free paths of acoustic phonons in the SiNC structures are equivalent or even below half the phonon wavelength, i.e., the minimum thermal conductivity scenario. The result demonstrates that the nanostructures with extremely small length scales and a controlled interface can give rise to ultimate classical confinement of thermal phonon propagation. read less USED (low confidence) B. M. Shenoy, G. Hegde, and D. Mahapatra, “Embedded silicon nanocrystal interface structure and strain,” Journal of Nanoparticle Research. 2018. link Times cited: 3 USED (low confidence) C. Fridlund, J. Laakso, K. Nordlund, and F. Djurabekova, “Atomistic simulation of ion irradiation of semiconductor heterostructures,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2017. link Times cited: 10 USED (low confidence) T. Li, Z. Tang, Z. Huang, and J. Yu, “Thermal boundary resistance between the polycrystalline graphene and the amorphous SiO 2 substrate,” Chemical Physics Letters. 2017. link Times cited: 12 USED (low confidence) N. Korobeishchikov, P. Stishenko, Y. Popenko, M. Roenko, and I. Nikolaev, “Interaction of accelerated argon cluster ions with a silicon dioxide surface.” 2017. link Times cited: 9 Abstract: Gas cluster ion beams bring new opportunities for diagnostic… read moreAbstract: Gas cluster ion beams bring new opportunities for diagnostics and modification of materials surfaces. In this work impact of argon clusters on silicon dioxide has been studied by molecular dynamics simulations and experimentally. We have obtained dependencies of crater size and the SiO2 sputtering yield on cluster size and specific energy. High reactive selectivity of sputtered products has been revealed for a high specific energy of clusters. It can cause modification of the target surface layer composition in case of long time irradiation. Peculiarities of experimental and computational data matching have been discussed.Gas cluster ion beams bring new opportunities for diagnostics and modification of materials surfaces. In this work impact of argon clusters on silicon dioxide has been studied by molecular dynamics simulations and experimentally. We have obtained dependencies of crater size and the SiO2 sputtering yield on cluster size and specific energy. High reactive selectivity of sputtered products has been revealed for a high specific energy of clusters. It can cause modification of the target surface layer composition in case of long time irradiation. Peculiarities of experimental and computational data matching have been discussed. read less USED (low confidence) S. Zhang et al., “Computational and Experimental Studies on Novel Materials for Fission Gas Capture.” 2017. link Times cited: 1 USED (low confidence) G. Wang et al., “Measuring Interlayer Shear Stress in Bilayer Graphene.,” Physical review letters. 2017. link Times cited: 151 Abstract: Monolayer two-dimensional (2D) crystals exhibit a host of in… read moreAbstract: Monolayer two-dimensional (2D) crystals exhibit a host of intriguing properties, but the most exciting applications may come from stacking them into multilayer structures. Interlayer and interfacial shear interactions could play a crucial role in the performance and reliability of these applications, but little is known about the key parameters controlling shear deformation across the layers and interfaces between 2D materials. Herein, we report the first measurement of the interlayer shear stress of bilayer graphene based on pressurized microscale bubble loading devices. We demonstrate continuous growth of an interlayer shear zone outside the bubble edge and extract an interlayer shear stress of 40 kPa based on a membrane analysis for bilayer graphene bubbles. Meanwhile, a much higher interfacial shear stress of 1.64 MPa was determined for monolayer graphene on a silicon oxide substrate. Our results not only provide insights into the interfacial shear responses of the thinnest structures possible, but also establish an experimental method for characterizing the fundamental interlayer shear properties of the emerging 2D materials for potential applications in multilayer systems. read less USED (low confidence) S. Suryavanshi, A. Gabourie, A. Farimani, E. Yalon, and E. Pop, “Thermal boundary conductance of the MOS2-SiO2 interface,” 2017 IEEE 17th International Conference on Nanotechnology (IEEE-NANO). 2017. link Times cited: 3 Abstract: We investigate heat conduction across the interface of a mon… read moreAbstract: We investigate heat conduction across the interface of a monolayer semiconductor and its supporting substrate using molecular dynamics (MD) simulations. For the first time, we show that for the interface between MoS2 and SiO2, thermal boundary conductance (TBC) is 15.5 ± 1.5 MWK −1m −2. The TBC is found to increase proportionally with the strength of the van der Waals interactions and is largely independent of temperature between 200 and 400 K. We also find that bi- and tri-layer MoS 2 on SiO 2 have somewhat higher TBC compared to single-layer MoS 2 on SiO 2. We compare the TBC simulation results with experimental data from Raman thermometry, finding close agreement between simulation and experiments. read less USED (low confidence) A. Sogoyan, D. Boychenko, and A. V. Demidova, “Simulation of impact of the HCP on the CNT-nanosensor by the molecular dynamics method,” Russian Microelectronics. 2017. link Times cited: 2 USED (low confidence) F. Meng, M. O. Elsahati, J. Liu, and R. Richards, “Thermal resistance between amorphous silica nanoparticles,” Journal of Applied Physics. 2017. link Times cited: 27 Abstract: Nanoparticle-based materials have been used as thermal insul… read moreAbstract: Nanoparticle-based materials have been used as thermal insulation in a variety of macroscale and microscale applications. In this work, we investigate the heat transfer between nanoparticles using non-equilibrium molecular dynamics simulations. We calculate the total thermal resistance and thermal boundary resistance between adjacent amorphous silica nanoparticles. Numerical results are compared to interparticle resistances determined from experimental measurements of heat transfer across packed silica nanoparticle beds. The thermal resistance between nanoparticles is shown to increase rapidly as the particle contact radius decreases. More significantly, the interparticle resistance depends strongly on the forces between particles, in particular, the presence or absence of chemical bonds between nanoparticles. In addition, the effect of interfacial force strength on thermal resistance increases as the nanoparticle diameter decreases. The simulations results are shown to be in good agreement with experimen... read less USED (low confidence) A. Sogoyan, D. Boychenko, and A. V. Demidova, “Simulation of impact of the HCP on the CNT-nanosensor by the molecular dynamics method,” Russian Microelectronics. 2017. link Times cited: 0 USED (low confidence) X. Zhang, S. Zhao, Y. Wang, and J. Xue, “Additivity of kinetic and potential energy contributions in modification of graphene supported on SiO2,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2017. link Times cited: 2 USED (low confidence) V. Kuryliuk and O. Korotchenkov, “Atomistic simulation of the thermal conductivity in amorphous SiO 2 matrix/Ge nanocrystal composites,” Physica E-low-dimensional Systems & Nanostructures. 2017. link Times cited: 5 USED (low confidence) Z. Liang, T. Wilson, and P. Keblinski, “Phonon interference in crystalline and amorphous confined nanoscopic films,” Journal of Applied Physics. 2017. link Times cited: 10 Abstract: Using molecular dynamics phonon wave packet simulations, we … read moreAbstract: Using molecular dynamics phonon wave packet simulations, we study phonon transmission across hexagonal (h)-BN and amorphous silica (a-SiO2) nanoscopic thin films sandwiched by two crystalline leads. Due to the phonon interference effect, the frequency-dependent phonon transmission coefficient in the case of the crystalline film (Si|h-BN|Al heterostructure) exhibits a strongly oscillatory behavior. In the case of the amorphous film (Si|a-SiO2|Al and Si|a-SiO2|Si heterostructures), in spite of structural disorder, the phonon transmission coefficient also exhibits oscillatory behavior at low frequencies (up to ∼1.2 THz), with a period of oscillation consistent with the prediction from the two-beam interference equation. Above 1.2 THz, however, the phonon interference effect is greatly weakened by the diffuse scattering of higher-frequency phonons within an a-SiO2 thin film and at the two interfaces confining the a-SiO2 thin film. read less USED (low confidence) N. Liao, M. Zhang, H. Zhou, and W. Xue, “Modeling of amorphous SiCxO6/5 by classical molecular dynamics and first principles calculations,” Scientific Reports. 2017. link Times cited: 5 USED (low confidence) S. Cao, H. He, and W. Zhu, “Defect induced phonon scattering for tuning the lattice thermal conductivity of SiO2 thin films,” AIP Advances. 2017. link Times cited: 4 Abstract: In this work, the thermal properties of nanoscale SiO2 thin … read moreAbstract: In this work, the thermal properties of nanoscale SiO2 thin films have been systematically investigated with respect to the thickness, crystal orientations and the void defects using non-equilibrium molecular-dynamics (NEMD) simulation. Size effect for the lattice thermal conductivity of nanoscale SiO2 thin films was observed. Additionally, SiO2 thin films with [001] oriented exhibited greater thermal conductivity compared with other crystal orientations which was discussed in terms of phonon density of states (PDOS). Furthermore, the porosity of void defects was introduced to quantify the influence of defects for thermal conductivity. Results exhibited that the thermal conductivity degraded with the increase of porosity. Two thermal conductivity suppression mechanisms, namely, void defects induced material loss interdicting heat conduction and phonon scattering enhanced by the boundary of defects, were proposed. Then, a further simulation was deployed to find that the effect of boundary scattering of def... read less USED (low confidence) N. Liao, B. Zheng, H. Zhou, and W. Xue, “Effect of carbon segregation on performance of inhomogeneous SiC y O 6/5 as anode materials for lithium-ion battery: A first-principles study,” Journal of Power Sources. 2016. link Times cited: 26 USED (low confidence) J. Li, E. Lampin, C. Delerue, and Y. Niquet, “Theoretical investigation of the phonon-limited carrier mobility in (001) Si films,” Journal of Applied Physics. 2016. link Times cited: 6 Abstract: We calculate the phonon-limited carrier mobility in (001) Si… read moreAbstract: We calculate the phonon-limited carrier mobility in (001) Si films with a fully atomistic framework based on a tight-binding (TB) model for the electronic structure, a valence-force-field model for the phonons, and the Boltzmann transport equation. This framework reproduces the electron and phonon bands over the whole first Brillouin zone and accounts for all possible carrier-phonon scattering processes. It can also handle one-dimensional (wires) and three-dimensional (bulk) structures and therefore provides a consistent description of the effects of dimensionality on the phonon-limited mobilities. We first discuss the dependence of the electron and hole mobilities on the film thickness and carrier density. The mobility tends to decrease with decreasing film thickness and increasing carrier density, as the structural and electric confinement enhances the electron-phonon interactions. We then compare hydrogen-passivated and oxidized films in order to understand the impact of surface passivation on the mobi... read less USED (low confidence) V. Kuryliuk and S. Semchuk, “Molecular Dynamics Calculation of Thermal Conductivity in a-SiO2 and an a-SiO2-Based Nanocomposite,” Ukrainian Journal of Physics. 2016. link Times cited: 3 Abstract: Thermal conductivity in amorphous SiO 2 ( 𝑎 -SiO 2 ) has be… read moreAbstract: Thermal conductivity in amorphous SiO 2 ( 𝑎 -SiO 2 ) has been studied in a wide range of temperatures, by using the nonequilibrium molecular dynamics method and the Beest–Kramer–Santen, Tersoff, and Vashishta empirical potentials. The thermal conductivity of an 𝑎 -SiO 2 -based composite with Si nanocrystals is calculated with the use of the Tersoff potential. The thermal conductivity of the nanocomposite is shown to firstly decrease and then to increase, as the silicon volumetric ratio grows. The obtained results are explained by the enhanced scattering of thermal vibrations at the matrix–Si nanocrystal boundaries. read less USED (low confidence) C. Liang et al., “Fractal nature of non-spherical silica particles via facile synthesis for the abrasive particles in chemical mechanical polishing,” Colloids and Surfaces A: Physicochemical and Engineering Aspects. 2016. link Times cited: 5 USED (low confidence) D. G. Kizzire et al., “Investigations of the Mechanical and Hydrothermal Stabilities of SBA-15 and Al-SBA-15 Mesoporous Materials,” MRS Advances. 2016. link Times cited: 3 Abstract: Periodic mesoporous materials possess high surface to volume… read moreAbstract: Periodic mesoporous materials possess high surface to volume ratio and nano-scale sized pores, making them potential candidates for heterogeneous catalysis, ion exchange, gas sensing and other applications. In this study, we use in situ small angle x-ray scattering (SAXS) and molecular dynamics (MD) simulations to investigate the mechanical and hydrothermal stability properties of periodic mesoporous SBA-15 silica and SBA-15 type aluminosilica (Al-SBA-15) to extreme conditions. The mesoporous SBA-15 silica and Al-SBA-15 aluminosilica possess amorphous frameworks and have similar pore size distribution (pore size ~9–10 nm). The in situ SAXS measurements were made at the B1 beamline, at the Cornell High Energy Synchrotron Source (CHESS). The mesoporous SBA-15 silica and Al-SBA-15 aluminosilica specimens were loaded in a diamond anvil cell (DAC) for pressure measurements, and, separately, with water in the DAC for hydrothermal measurements to high P-T conditions (to 255 °C and ~ 114 MPa). Analyses of the pressure-dependent SAXS data show that the mesoporous Al-SBA-15 aluminosilica is substantially more mechanically stable than the SBA-15 silica. Hydrothermal measurements show a small net swelling of the framework at elevated P-T conditions, due to dissolution of water into the pore walls. Under elevated P-T conditions, the Al-SBA-15 aluminosilica shows significantly greater hydrothermal stability than the SBA-15 silica. Our MD simulations show that the bulk modulus value of periodic mesoporous SBA-15 silica varies exponentially with percentage porosity. Molecular dynamics simulations are being made in order to better understand how the pore architecture and the chemical composition of the host structure govern the stability properties of the mesoporous materials. read less USED (low confidence) C. Shao and H. Bao, “Thermal transport in bismuth telluride quintuple layer: mode-resolved phonon properties and substrate effects,” Scientific Reports. 2016. link Times cited: 25 USED (low confidence) S. Mann and P. Rani, “Study of interaction in silica glass via model potential approach.” 2016. link Times cited: 0 Abstract: Silica is one of the most commonly encountered substances in… read moreAbstract: Silica is one of the most commonly encountered substances in daily life and in electronics industry. Crystalline SiO2 (in several forms: quartz, cristobalite, tridymite) is an important constituent of many minerals and gemstones, both in pure form and mixed with related oxides. Cohesive energy of amorphous SiO2 has been investigated via intermolecular potentials i.e weak Van der Waals interaction and Morse type short-range interaction.We suggest a simple atom-atom based Van der Waals as well as Morse potential to find cohesive energy of glass. It has been found that the study of silica structure using two different model potentials is significantly different. Van der Waals potential is too weak (P.E =0.142eV/molecule) to describe the interaction between silica molecules. Morse potential is a strong potential, earlier given for intramolecular bonding, but if applied for intermolecular bonding, it gives a value of P.E (=−21.92eV/molecule) to appropriately describe the structure of silica. read less USED (low confidence) B. Cowen and M. El-Genk, “Probability-based threshold displacement energies for oxygen and silicon atoms in α-quartz silica,” Computational Materials Science. 2016. link Times cited: 17 USED (low confidence) J. Zhen et al., “Molecular dynamics study of structural damage in amorphous silica induced by swift heavy-ion radiation,” Radiation Effects and Defects in Solids. 2016. link Times cited: 16 Abstract: ABSTRACT In this paper, the radiation defects induced by the… read moreAbstract: ABSTRACT In this paper, the radiation defects induced by the swift heavy ions and the recoil atoms in amorphous SiO2 were studied. The energy of recoil atoms induced by the incident Au ions in SiO2 was calculated by using Monte Carlo method. Results show that the average energies of recoils reach the maximum (200 eV for Si and 130 eV for O, respectively) when the incident energy of Au ion is 100 MeV. Using Tersoff/zbl potential with the newly built parameters, the defects formation processes in SiO2 induced by the recoils were studied by using molecular dynamics method. The displacement threshold energies (Ed) for Si and O atoms are found to be 33.5 and 16.3 eV, respectively. Several types of under- and over-coordinated Si and O defects were analyzed. The results demonstrate that Si3, Si5, and O1 are the mainly defects in SiO2 after radiation. Besides, the size of cylindrical damage region produced by a single recoil atom was calculated. The calculation shows that the depth and the radius are up to 2.0 and 1.4 nm when the energy of recoils is 200 eV. Finally, it is estimated that the Au ion would induce a defected track with a diameter of 4 nm in SiO2. read less USED (low confidence) S. Zhao and J. Xue, “Modification of graphene supported on SiO2 substrate with swift heavy ions from atomistic simulation point,” Carbon. 2015. link Times cited: 41 USED (low confidence) I. Fadhilah and Y. Rosandi, “Molecular-dynamics study of amorphous SiO2 relaxation.” 2015. link Times cited: 1 Abstract: Using Molecular-Dynamics simulation we observed the generati… read moreAbstract: Using Molecular-Dynamics simulation we observed the generation of amorphous SiO2 target from a randomly distributed Si and O atoms. We applied a sequence of annealing of the target with various temperature and quenching to room temperature. The relaxation time required by the system to form SiO4 tetrahedral mesh after a relatively long simulation time, is studied. The final amorphous target was analyzed using the radial distribution function method, which can be compared with the available theoretical and experimental data. We found that up to 70% of the target atoms form the tetrahedral SiO4 molecules. The number of formed tetrahedral increases following the growth function and the rate of SiO4 formation follows Arrhenius law, depends on the annealing temperature. The local structure of amorphous SiO2 after this treatment agrees well with those reported in some literatures. read less USED (low confidence) F. Grigoriev, A. Sulimov, I. Kochikov, O. Kondakova, V. Sulimov, and A. Tikhonravov, “Supercomputer modeling of the ion beam sputtering process: full-atomistic level,” SPIE Optical Systems Design. 2015. link Times cited: 18 Abstract: A new method for supercomputer atomistic modeling of the ion… read moreAbstract: A new method for supercomputer atomistic modeling of the ion beam sputtering process is presented allowing atomistic modeling of the systems consisting of 106 – 108 atoms. Deposition process is organized as a sequence of molecular dynamic cycles in which deposited atoms interact with the substrate with earlier deposited atoms and form new chemical bonds. The method is applied to the modeling of SiO2 thin optical films. For interatomic potential energy calculation the original DESIL force field with high computational efficiency has been developed. Atomistic modeling of the deposition processes with different Si atom energies is performed for the films with thicknesses up to 30 nm (about one million deposited atoms). Dependence of thin film density on film thickness is investigated. It is found that film densities depend on the energy of sputtered atoms and exceed the density of fused silica substrate by 0.1-0.2 g/cm3. In all experiments interface layers with the thicknesses of about 1-2 nm between thin film and substrate are observed. read less USED (low confidence) B. Cowen and M. El-Genk, “On force fields for molecular dynamics simulations of crystalline silica,” Computational Materials Science. 2015. link Times cited: 31 USED (low confidence) D. Berman, S. A. Deshmukh, S. Sankaranarayanan, A. Erdemir, and A. Sumant, “Macroscale superlubricity enabled by graphene nanoscroll formation,” Science. 2015. link Times cited: 624 Abstract: Slip sliding away Many applications would benefit from ultra… read moreAbstract: Slip sliding away Many applications would benefit from ultralow friction conditions to minimize wear on the moving parts such as in hard drives or engines. On the very small scale, ultralow friction has been observed with graphite as a lubricant. Berman et al. achieved superlubricity using graphene in combination with crystalline diamond nanoparticles and diamondlike carbon (see the Perspective by Hone and Carpick). Simulations showed that sliding of the graphene patches around the tiny nanodiamond particles led to nanoscrolls with reduced contact area that slide easily against the amorphous diamondlike carbon surface. Science, this issue p. 1118; see also p. 1087 Nanodiamonds wrapped with graphene sheets lead to ultralow friction against a diamondlike carbon surface. [Also see Perspective by Hone and Carpick] Friction and wear remain as the primary modes of mechanical energy dissipation in moving mechanical assemblies; thus, it is desirable to minimize friction in a number of applications. We demonstrate that superlubricity can be realized at engineering scale when graphene is used in combination with nanodiamond particles and diamondlike carbon (DLC). Macroscopic superlubricity originates because graphene patches at a sliding interface wrap around nanodiamonds to form nanoscrolls with reduced contact area that slide against the DLC surface, achieving an incommensurate contact and substantially reduced coefficient of friction (~0.004). Atomistic simulations elucidate the overall mechanism and mesoscopic link bridging the nanoscale mechanics and macroscopic experimental observations. read less USED (low confidence) B. Gueye, Y. Zhang, Y. Wang, and Y. Chen, “Origin of frictional ageing by molecular dynamics simulation of a silicon tip sliding over a diamond substrate,” Tribology International. 2015. link Times cited: 6 USED (low confidence) C. Shao and H. Bao, “A molecular dynamics investigation of heat transfer across a disordered thin film,” International Journal of Heat and Mass Transfer. 2015. link Times cited: 27 USED (low confidence) F. Grigoriev, A. Sulimov, I. Kochikov, O. Kondakova, V. Sulimov, and A. Tikhonravov, “High-performance atomistic modeling of optical thin films deposited by energetic processes,” The International Journal of High Performance Computing Applications. 2015. link Times cited: 22 Abstract: In this paper we present a computationally effective approac… read moreAbstract: In this paper we present a computationally effective approach to classical molecular dynamic simulation of thin film growth with orientation on cluster supercomputing facilities. The goal of the developed approach is to investigate structural heterogeneities of thin films deposited on substrates at a nanoscale level. These heterogeneities depend on the experimental conditions of a deposition process being used. They have essential influence on practical properties of thin films and their modeling is important for achieving further progress in thin film optical technology. The presented research is focused on silicon dioxide thin films growth. A special force field, oriented on the atomistic description of the silicon dioxide deposition on fused silica substrate, has been developed and applied to the molecular dynamic simulation with the GROMACS package. The validity of the developed simulation approach is verified using atomic clusters consisting of up to 106 atoms and having characteristic dimensions of up to 30 nm. Its computational efficiency is tested using up to 2048 cores. The dependence of achievable efficiency on model parameters is discussed. read less USED (low confidence) A. Leino, S. Daraszewicz, O. Pakarinen, K. Nordlund, and F. Djurabekova, “Atomistic two-temperature modelling of ion track formation in silicon dioxide,” Europhysics Letters. 2015. link Times cited: 24 Abstract: We study swift-heavy-ion track formation in α-quartz using t… read moreAbstract: We study swift-heavy-ion track formation in α-quartz using the two-temperature molecular dynamics (2T-MD) model realised as a concurrent multiscale scheme. We compare the simulated track radii to the existing experimental ones obtained from small-angle X-ray scattering and Rutherford backscattering experiments. The 2T-MD model provides an explanation of the origin of the track radii saturation at high electronic stopping power. Furthermore, we study the track structure and show that defects formed outside the region of density fluctuations after a swift-heavy-ion impact may explain the conflicting track radii produced by the two experimental techniques. read less USED (low confidence) X. Hu, S. Sundararajan, and A. Martini, “The effects of adhesive strength and load on material transfer in nanoscale wear,” Computational Materials Science. 2014. link Times cited: 19 USED (low confidence) A. Ilinov and A. Kuronen, “Size-dependent elastic properties of oxidized silicon nanorods,” Journal of Applied Physics. 2014. link Times cited: 1 Abstract: In this work, we have simulated a three point bending test f… read moreAbstract: In this work, we have simulated a three point bending test for Si nanorods of different sizes with an oxide coating of different thicknesses using molecular dynamics simulations and the finite element modeling (FEM). We tested nanorods with diameters from 6 to 16 nm, which had lengths from 31 to 62 nm. Our aim was to estimate how well the elastic properties of Si nanorods can be described using the classical continuum mechanics approach. The agreement between the MD simulations and the FEM calculations was consistent for the pristine Si nanorods of all sizes, whereas the oxidized Si nanorods with small length-to-diameter ratio had much smaller effective bending moduli values than predicted by FEM. Our assumption is that it is due to the significant decrease of the shear modulus in the oxide layer of the smallest nanorods. We had also introduced surface stresses into the FEM models and found that their influence on the bending properties is more important for partially oxidized nanorods. read less USED (low confidence) Y. Zhang, Y. Li, Z. Wang, and K. Zhao, “Lithiation of SiO2 in Li-ion batteries: in situ transmission electron microscopy experiments and theoretical studies.,” Nano letters. 2014. link Times cited: 115 Abstract: Surface passivation has become a routine strategy of design … read moreAbstract: Surface passivation has become a routine strategy of design to mitigate the chemomechanical degradation of high-capacity electrodes by regulating the electrochemical process of lithiation and managing the associated deformation dynamics. Oxides are the prevalent materials used for surface coating. Lithiation of SiO2 leads to drastic changes in its electro-chemo-mechanical properties from an electronic insulator and a brittle material in its pure form to a conductor and a material sustainable of large deformation in the lithiated form. We synthesized SiO2-coated SiC nanowires that allow us to focus on the lithiation behavior of the sub-10 nm SiO2 thin coating. We systematically investigate the structural evolution, the electronic conduction and ionic transport properties, and the deformation pattern of lithiated SiO2 through coordinated in situ transmission electron microcopy experiments, first-principles computation, and continuum theories. We observe the stress-mediated reaction that induces inhomogeneous growth of SiO2. The results provide fundamental perspectives on the chemomechanical behaviors of oxides used in the surface coating of Li-ion technologies. read less USED (low confidence) O. Malyi, V. V. Kulish, and C. Persson, “In search of new reconstructions of (001) α-quartz surface: a first principles study,” RSC Advances. 2014. link Times cited: 22 Abstract: Using Born–Oppenheimer molecular dynamics (BOMD) simulations… read moreAbstract: Using Born–Oppenheimer molecular dynamics (BOMD) simulations and “static” density functional theory (DFT) calculations, the stability of cleaved and reconstructed α-SiO2(001) surfaces was studied. We found reconstructions (“dense”, 2 × 2 reoptimized “dense”, and 3 × 3 reoptimized “dense”) which minimize the surface energy. The analysis of the surface energies shows that the cleaved surface reconstructs to the 2 × 2 reoptimized “dense” surface having a surface energy around 10% smaller than the “dense” surface. The results suggest that the optimization of Si–Si and Si–O distances at top surface layers plays the key role in stabilizing the 2 × 2 “dense” surface over the well-known “dense” surface. read less USED (low confidence) N. Liao, W. Xue, H. Zhou, and M. Zhang, “Molecular dynamics investigation of structure and high-temperature mechanical properties of SiBCO ceramics,” Journal of Alloys and Compounds. 2014. link Times cited: 16 USED (low confidence) H. Minari et al., “Defect formation in III–V fin grown by aspect ratio trapping technique: A first-principles study,” 2014 IEEE International Reliability Physics Symposium. 2014. link Times cited: 0 Abstract: First-principles investigations are used to study the format… read moreAbstract: First-principles investigations are used to study the formation of defects in III-V fins grown using the aspect ratio trapping technique. We show that, during the growth of the III-V, the formation of intermediate chemical states with the precursors leads to the creation of in-diffused Mg/Zn and Al2O3 sub-oxide. Our prediction is consistent with the experimental observations. These defect formations could be at the origin of the degradation of the electrical reliability of III-V fin-shaped field-effect transistors and the cause of the increasing difficulties met in the fabrication of III-V fin. read less USED (low confidence) R. Su, M. Xiang, J. Chen, S. Jiang, and H. Wei, “Molecular dynamics simulation of shock induced ejection on fused silica surface,” Journal of Applied Physics. 2014. link Times cited: 21 Abstract: Shock response and surface ejection behaviors of fused silic… read moreAbstract: Shock response and surface ejection behaviors of fused silica are studied by using non-equilibrium molecular dynamics combining with the Tersoff potential. First, bulk structure and Hugoniot curves of fused silica are calculated and compared with experimental results. Then, the dynamical process of surface ejection behavior is simulated under different loading velocities ranging from 3.5 to 5.0 km∕s, corresponding to shock wave velocities from 7.1 to 8.8 km∕s. The local atomistic shear strain parameter is used to describe the local plastic deformation under conditions of shock compression or releasing. Our result shows that the shear strain is localized in the bottom area of groove under the shock compression. Surface ejection is observed when the loading velocity exceeds 4.0 km∕s. Meanwhile, the temperature of the micro-jet is ∼5574.7 K, which is close to experiment measurement. Several kinds of structural defects including non-bridging oxygen are found in the bulk area of the sample after ejection. read less USED (low confidence) A. Ilinov and A. Kuronen, “Atomistic modeling of bending properties of oxidized silicon nanowires,” Journal of Applied Physics. 2014. link Times cited: 8 Abstract: In this work, we have modeled a three point bending test of … read moreAbstract: In this work, we have modeled a three point bending test of monocrystalline Si nanowires using molecular dynamics simulations in order to investigate their elastic properties. Tested nanowires were about 30 nm in length and had diameters from 5 to 9 nm. To study the influence of a native oxide layer, nanowires were covered with a 1 nm thick silica layer. The bending force was applied by a carbon diamond half-sphere with a 5 nm diameter. The Si-O parametrization for the Tersoff potential was used to describe atomic interactions between Si and O atoms. In order to remove the indentation effect of the diamond half-sphere and to obtain a pure bending behavior, we have also performed a set of simulations with fixed bottoms of the nanowires. Our results show that the oxide layer reduces the nanowire stiffness when compared with a pure Si nanowire with the same number of silicon atoms—in spite of the fact that the oxidized nanowires had larger diameters. read less USED (low confidence) Y. Ding, Z. Guo, X. Su, and Y. Zhang, “Molecular Dynamics Simulation of Void Structures in Vitreous Silica,” Advanced Materials Research. 2014. link Times cited: 0 Abstract: In order to investigate the density anomaly of vitreous sili… read moreAbstract: In order to investigate the density anomaly of vitreous silica in the medium-or long-range order, different models were made to study the atomic configuration revolution in thermal history by molecular dynamics. The void structures have been studied through analyzing the best model that is carefully selected. The principle of void size distribution revolution at elevated temperatures was used to explain the density anomaly of the vitreous silica. The simulation results showed that when the temperature is low, the void radius increases with the temperature rising. After 2000 K, large void structures are destroyed, filled, or separated into small radius voids. In the range from 2000 K to 2400 K, large void structures decrease faster exceeding the bond extension on thermal expansion contribution, this should be the root cause of negative thermal expansion behavior for vitreous silica. When the temperature is greater than 2400 K, with the temperature rising, the normal thermal expansion is recovered gradually because number of large voids has been reduced and their destroying cannot eliminate the contribution to expansion of bond extension. Therefore, the negative thermal expansion of vitreous silica could be described by the revolution of void structures in the medium-or long-range clearly, and is mainly influenced by the existence and change of larger voids. read less USED (low confidence) P. Y. Huang et al., “Imaging Atomic Rearrangements in Two-Dimensional Silica Glass: Watching Silica’s Dance,” Science. 2013. link Times cited: 205 Abstract: Glassy Eyed In crystalline materials, the collective motion … read moreAbstract: Glassy Eyed In crystalline materials, the collective motion of atoms in one- and two-dimensional defects—like dislocations and stacking faults—controls the response to an applied strain, but how glassy materials change their structure in response to strain is much less clear. Huang et al. (p. 224; see the Perspective by Heyde) used advanced-transmission electron microscopy to investigate the structural rearrangements in a two-dimensional glass, including the basis for shear deformations and the atomic behavior at the glass/liquid interface. Dynamics of individual atoms in a two-dimensional silicate glass have been observed using transmission electron microscopy. [Also see Perspective by Heyde] Structural rearrangements control a wide range of behavior in amorphous materials, and visualizing these atomic-scale rearrangements is critical for developing and refining models for how glasses bend, break, and melt. It is difficult, however, to directly image atomic motion in disordered solids. We demonstrate that using aberration-corrected transmission electron microscopy, we can excite and image atomic rearrangements in a two-dimensional silica glass—revealing a complex dance of elastic and plastic deformations, phase transitions, and their interplay. We identified the strain associated with individual ring rearrangements, observed the role of vacancies in shear deformation, and quantified fluctuations at a glass/liquid interface. These examples illustrate the wide-ranging and fundamental materials physics that can now be studied at atomic-resolution via transmission electron microscopy of two-dimensional glasses. read less USED (low confidence) S. D. Nath, “Study of the effect of sizes on the structural properties of SiO2 glass by molecular dynamics simulations,” Journal of Non-crystalline Solids. 2013. link Times cited: 8 USED (low confidence) K. Kleovoulou and P. Kelires, “Stress state of embedded Si nanocrystals,” Physical Review B. 2013. link Times cited: 19 USED (low confidence) C. Y. Chuang, Q. Li, D. Leonhardt, S. Han, and T. Sinno, “Atomistic analysis of Ge on amorphous SiO2 using an empirical interatomic potential,” Surface Science. 2013. link Times cited: 14 USED (low confidence) T. Blue, W. Windl, and B. Dickerson, “Testing of Performance of Optical Fibers Under Irradiation in Intense Radiation Fields, When Subjected to Very High Temperatures.” 2013. link Times cited: 3 Abstract: The primary objective of this project is to measure and mode… read moreAbstract: The primary objective of this project is to measure and model the performance of optical fibers in intense radiation fields when subjected to very high temperatures. This research will pave the way for fiber optic and optically based sensors under conditions expected in future high-temperature gas-cooled reactors. Sensor life and signal-to-noise ratios are susceptible to attenuation of the light signal due to scattering and absorbance in the fibers. This project will provide an experimental and theoretical study of the darkening of optical fibers in high-radiation and high-temperature environments. Although optical fibers have been studied for moderate radiation fluence and flux levels, the results of irradiation at very high temperatures have not been published for extended in-core exposures. Several previous multi-scale modeling efforts have studied irradiation effects on the mechanical properties of materials. However, model-based prediction of irradiation-induced changes in silicaA¢ÂÂs optical transport properties has only recently started to receive attention due to possible applications as optical transmission components in fusion reactors. Nearly all damage-modeling studies have been performed in the molecular-dynamics domain, limited to very short times and small systems. Extended-time modeling, however, is crucial to predicting the long-term effects of irradiation at high temperatures, since the experimental testing may not encompass the displacement rate that the fibers will encounter if they are deployed in the VHTR. The more » project team will pursue such extended-time modeling, including the effects of the ambient and recrystallization. The process will be based on kinetic MC modeling using the concept of amorphous material consisting of building blocks of defect-pairs or clusters, which has been successfully applied to kinetic modeling in amorphized and recrystallized silicon. Using this procedure, the team will model compensation for rate effects, and the interplay of rate effects with the effects of annealing, to accurately predict the fibersA¢Â reliability and expected lifetime « less read less USED (low confidence) J. Chen, G. Zhang, and B. Li, “Substrate coupling suppresses size dependence of thermal conductivity in supported graphene.,” Nanoscale. 2013. link Times cited: 5 Abstract: Thermal conductivity κ of both suspended and supported graph… read moreAbstract: Thermal conductivity κ of both suspended and supported graphene has been studied by using molecular dynamics simulations. An obvious length dependence is observed in κ of suspended single-layer graphene (SLG), while κ of supported SLG is insensitive to the length. The simulation result of room temperature κ of supported SLG is in good agreement with the experimental value. In contrast to the decrease in κ induced by inter-layer interaction in suspended few-layer graphene (FLG), κ of supported FLG is found to increase rapidly with the layer thickness, reaching about 90% of that of bulk graphite at six layers, and eventually saturates at the thickness of 13.4 nm. More interestingly, unlike the remarkable substrate dependent κ in SLG, the effect of substrate on thermal transport is much weaker in FLG. The underlying physics is investigated and presented. read less USED (low confidence) N. Liao, “Large-Scale Molecular Dynamics Modeling of a-SiO2,” Advanced Materials Research. 2012. link Times cited: 0 Abstract: Silicon dioxide plays an important role in integrated circui… read moreAbstract: Silicon dioxide plays an important role in integrated circuits and microelectronics. However, the experiments have limitations in micro/nano-scale characterization of fracture properties at high temperatures. In this paper, the structural and fracture properties of amorphous silicon dioxide (a-SiO2) were studied at temperatures up to 1500K. The simulation results consist with the experiments on pair distribution functions, structure factor and angular distributions. read less USED (low confidence) A. Galashev, “A computer study of the Raman spectra of the (GaN)129, (SiO2)86, and (GaN)54(SiO2)50 nanoparticles,” Russian Journal of Physical Chemistry B. 2012. link Times cited: 2 USED (low confidence) B. Qiu, Y. Wang, Q. Zhao, and X. Ruan, “The effects of diameter and chirality on the thermal transport in free-standing and supported carbon-nanotubes,” Applied Physics Letters. 2012. link Times cited: 43 Abstract: We use molecular dynamics simulations to explore the lattice… read moreAbstract: We use molecular dynamics simulations to explore the lattice thermal transport in free-standing and supported single-wall carbon-nanotube (SWCNT) in comparison to that in graphene nanoribbon and graphene sheet. For free-standing SWCNT, the lattice thermal conductivity increases with diameter and approaches that of graphene, partly due to the curvature. Supported SWCNT thermal conductivity is reduced by 34%-41% compared to the free-standing case, which is less than that in supported graphene. Also, it shows an evident chirality dependence by varying about 10%, which we attribute to chirality-dependent interfacial phonon scattering. read less USED (low confidence) G. Balasubramanian, S. Sen, and I. Puri, “Shear viscosity enhancement in water–nanoparticle suspensions,” Physics Letters A. 2012. link Times cited: 14 USED (low confidence) Y. He, Y.-Z. Tang, M. Ding, and L. Ma, “Thermal Conductivity of Amorphous and Crystalline SiO2 Nano-Films from Molecular Dynamics Simulations,” Key Engineering Materials. 2012. link Times cited: 1 Abstract: Normal thermal conductivity of amorphous and crystalline SiO… read moreAbstract: Normal thermal conductivity of amorphous and crystalline SiO2 nano-films is calculated by nonequilibrium molecular dynamics (NEMD) simulations in the temperature range from 100 to 700K and thicknesses from 2 to 6nm. The calculated temperature and thickness dependences of thermal conductivity are in good agreement with previous literatures. In the same thickness, higher thermal conductivity is obtained for crystalline SiO2 nano-films. And more importantly, for amorphous SiO2 nano-films, thickness can be any direction of x, y, z-axis without effect on the normal thermal conductivity, for crystalline SiO2 nano-films, the different thickness directions obtain different thermal conductivity results. The different results of amorphous and crystalline SiO2 nano-films simply show that film thickness and grain morphology will cause different effects on thermal conductivity. read less USED (low confidence) B. Qiu and X. Ruan, “Molecular dynamics simulations of thermal conductivity and spectral phonon relaxation time in suspended and supported graphene,” arXiv: Mesoscale and Nanoscale Physics. 2011. link Times cited: 148 Abstract: We perform molecular dynamics (MD) simulations with phonon s… read moreAbstract: We perform molecular dynamics (MD) simulations with phonon spectral analysis aiming at understanding the two dimensional (2D) thermal transport in suspended and supported graphene. Within the framework of equilibrium MD simulations, we perform spectral energy density (SED) analysis to obtain the lifetime of individual phonon modes. The per-mode contribution to thermal conductivity is then calculated to obtain the lattice thermal conductivity in the temperature range 300-650 K. In contrast to prior studies, our results suggest that the contribution from out-of-plane acoustic (or ZA) branch to thermal conductivity is around 25-30% in suspended single-layer graphene (SLG) at room temperature. The thermal conductivity is found to reduce when SLG is put on amorphous SiO2 substrate. Such reduction is attributed to the strengthened scattering in all phonon modes in the presence of the substrate. Among them, ZA modes are mostly affected with their contribution to thermal conductivity reduced to around 15%. As a result, thermal transport is dominated by in-plane acoustic phonon modes in supported SLG. read less USED (low confidence) M. Z. Hossain, “Semiconducting graphene nanoribbon retains band gap on amorphous or crystalline SiO2,” Applied Physics Letters. 2011. link Times cited: 1 Abstract: Electronic properties of a semiconducting armchair graphene … read moreAbstract: Electronic properties of a semiconducting armchair graphene nanoribbon on SiO_2 are examined using first-principles calculations and taking into account the van der Waals interaction. Unlike semiconducting carbon nanotubes, which exhibit variations in band gap on SiO_2, the nanoribbon is
found to retain its band gap on SiO_2, regardless of the separation distance or the dielectric’s surface type—crystalline or amorphous. The interfacial interaction leads to electron-transfer from the nanoribbon to the dielectric. Moreover, for crystalline SiO_2, the quantity of electron-transfer and the binding energy depend strongly on the type of surface termination and weakly on the binding
sites. read less USED (low confidence) C.-J. Huang, C.-J. Wu, H. Teng, and K. Chiang, “A robust nano-mechanics approach for tensile and modal analysis using atomistic–continuum mechanics method,” Computational Materials Science. 2011. link Times cited: 1 USED (low confidence) J. Fan, “Applications of Atomistic Simulation in Ceramics and Metals.” 2010. link Times cited: 0 USED (low confidence) Z. Ong and E. Pop, “Frequency and polarization dependence of thermal coupling between carbon nanotubes and SiO2,” Journal of Applied Physics. 2010. link Times cited: 38 Abstract: We study heat dissipation from a (10,10) carbon nanotube (CN… read moreAbstract: We study heat dissipation from a (10,10) carbon nanotube (CNT) to a SiO2 substrate using equilibrium and nonequilibrium classical molecular dynamics. The CNT-substrate thermal boundary conductance is computed both from the relaxation time of the CNT-substrate temperature difference, and from the time autocorrelation function of the interfacial heat flux at equilibrium (Green–Kubo relation). The power spectrum of interfacial heat flux fluctuation and the time evolution of the internal CNT energy distribution suggest that: (1) thermal coupling is dominated by long wavelength phonons between 0–10 THz, (2) high frequency (40–57 THz) CNT phonon modes are strongly coupled to sub-40 THz CNT phonon modes, and (3) inelastic scattering between the CNT phonons and substrate phonons contributes to interfacial thermal transport. We also find that the low frequency longitudinal acoustic and twisting acoustic modes do not transfer energy to the substrate as efficiently as the low frequency transverse optical mode. read less USED (low confidence) O. Okeke and J. Lowther, “Molecular dynamics of binary metal nitrides and ternary oxynitrides,” Physica B-condensed Matter. 2009. link Times cited: 5 USED (low confidence) S. Tyaginov et al., “Description of Si-O bond breakage using pair-wise interatomic potentials under consideration of the whole crystal,” 2009 IEEE International Reliability Physics Symposium. 2009. link Times cited: 0 Abstract: We extend the McPherson model in a manner to capture the eff… read moreAbstract: We extend the McPherson model in a manner to capture the effect of the whole surrounding lattice on the siliconoxygen bond-breakage energetics. It is shown that the Mie- Grüneisen potential with the constants used in the original version of the model is not suitable under the consideration of the whole crystal. Other empirical pair-wise interatomic potentials, namely TTAM and BKS have been tested for the analysis of the bond rupture energetics. It is shown that the secondary minimum corresponding to the transition of the Si atom from the 4-fold to the 3-fold coordinated position occurs in a different direction with a rather high activation energy (~ 6 eV). The tunneling of the Si ion between the primary and the secondary minima has been treated within the WKB approximation. We demonstrate that the contribution of neighboring SiO4 tetrahedrons substantially decreases the breakage rate, making bond rupture by means of an electric field alone practically impossible. Therefore, the common action of an electric field and another contribution (bond weakening by hole capture, structural disorder and energy deposited by particles) is essential for Si-O bond-breakage. read less USED (low confidence) W. Malfait, W. Halter, and R. Verel, “29Si NMR spectroscopy of silica glass: T1 relaxation and constraints on the Si–O–Si bond angle distribution,” Chemical Geology. 2008. link Times cited: 67 USED (low confidence) B.-M. Lee, T. Motooka, and S. Munetoh, “Molecular-dynamics simulations of nucleation and crystallization processes of laser crystallized poly-Si,” Journal of Physics: Condensed Matter. 2008. link Times cited: 8 Abstract: The nucleation and crystallization processes of excimer-lase… read moreAbstract: The nucleation and crystallization processes of excimer-laser annealed Si on a SiO2 substrate for complete melting conditions have been investigated by using molecular-dynamics simulations. In the early stage of nucleation, the preferential growth of nuclei with a {111} face normal to the surface was originated from the {111} twin boundaries with a low surface energy. The partial rotation of the dimer leads to the growth of {111}-oriented nuclei along twins that have different stacking sequences. The recombination of vacancies and dimers at the solidification front is directly related to {111} growth from the twin boundaries. read less USED (low confidence) I. Nikolaev, P. Stishenko, N. G. Korobeishchikov, and V. Yakovlev, “Simulation of surface sputtering of fused quartz by clusters of different gases,” OIL AND GAS ENGINEERING (OGE-2022). 2023. link Times cited: 0 USED (low confidence) V.-T. Nguyen and T. Fang, “Revealing the mechanisms for inactive rolling and wear behaviour on chemical mechanical planarization,” Applied Surface Science. 2022. link Times cited: 7 USED (low confidence) O. Kaya and N. Donmezer, “Investigation of the Thermal Transport Properties Across Van der Waals Interfaces of 2D Materials,” IEEE Transactions on Nanotechnology. 2022. link Times cited: 0 Abstract: Two-dimensional (2D) materials have attracted extensive rese… read moreAbstract: Two-dimensional (2D) materials have attracted extensive research interest in various applications in recent years due to their superior thermal, electrical, and optical properties, making them preferable for potential electronic and optoelectronic applications. These 2D materials form Van der Waals interfaces with common substrate materials due to fabrication and/or device requirements. Since the generated heat during the operation of the devices cause degradation and reliability concerns, interface thermal boundary conductance (TBCs) and in-plane thermal conductivities of the interfaces should be well understood for proper thermal management. Herein, we investigate the TBC and in-plane thermal conductivities of the Van der Waals interfaces of 2D materials by approach to-equilibrium molecular dynamics (AEMD) and non-equilibrium molecular dynamics (NEMD) simulations. Our results show that the TBC is higher for the interfaces with stronger phonon DOS and lattice match. Also, the increasing number of 2D material layers increases the TBC of the interface. The results also showed that the thermal conductivity of the materials forming the interface could affect each other's in-plane thermal conductivity. Changes in thermal conductivities of individual in-plane thermal conductivities can be as high as 70%. Change in thermal conductivity depends on the difference in thermal conductivities of materials in contact and only visible in the vicinity of the interface. Thermal management strategies should pay attention to the trade-off between the changes in individual thermal conductivities and TBC of the interfaces. read less USED (low confidence) P. I. Chernovol and A. Mirzoev, “MOLECULAR DYNAMIC MODELING OF STRUCTURE AND PROPERTIES OF SIO2 GLASS SUBSTRATE IN A BROAD TEMPERATURE RANGE,” Bulletin of the South Ural State University series "Mathematics. Mechanics. Physics". 2022. link Times cited: 0 Abstract: F.V. Grigoriev's paper “Force Fields for Molecular-Dyna… read moreAbstract: F.V. Grigoriev's paper “Force Fields for Molecular-Dynamic Modeling of Sputter Deposition Process on Silicon Dioxide Film”, proposes a simple DESIL inter-ion potential for modeling amorphous SiO2 substrates widely used for thin film sputter deposition. This potential provides an important advantage when compared with popular potential of BKS due to the absence of a non-physical attraction region at short distances between particles. This is important when simulating SiO2 substrate sputtering where collisions between particles result in short-range proximity. In this case an artifact of particle inter-capture may be observed which distorts the simulation results. The purpose of this work is to demonstrate the potential for predicting the structural and thermodynamic characteristics of amorphous silicate glasses over a broad temperature range. read less USED (low confidence) K. Yang, Q. Yang, X. Zhu, W. Hui, Z. Tao, and J. Liu, “A molecular dynamics simulation on the static calibration test of a revised thin-film thermopile heat-flux sensor,” Measurement. 2020. link Times cited: 8 USED (low confidence) K. Nordlund and F. Djurabekova, “Molecular Dynamics Simulations of Non-equilibrium Systems,” Handbook of Materials Modeling. 2020. link Times cited: 3 USED (low confidence) J. Yeo, Z. Liu, and T. Ng, “Silica Aerogels: A Review of Molecular Dynamics Modelling and Characterization of the Structural, Thermal, and Mechanical Properties,” Handbook of Materials Modeling. 2020. link Times cited: 9 USED (low confidence) N. A. Mehta, A. Rayabharam, and D. Levin, “Simulations of surface evolution due to particulate-surface interaction,” AIAA Scitech 2019 Forum. 2019. link Times cited: 2 USED (low confidence) F. Mu et al., “Room Temperature SiC-SiO2Wafer Bonding Enhanced by Using an Intermediate Si Nano Layer,” ECS Journal of Solid State Science and Technology. 2017. link Times cited: 9 USED (low confidence) B. Cowen and M. El-Genk, “Bond-order reactive force fields for molecular dynamics simulations of crystalline silica,” Computational Materials Science. 2016. link Times cited: 15 USED (low confidence) E. Lampin, “Recrystallization of Silicon by Classical Molecular Dynamics.” 2015. link Times cited: 0 USED (low confidence) Z. Ong and E. Pop, “Molecular Dynamics Simulation of Interfacial Thermal Resistance Between a (10,10) Carbon Nanotube and SiO 2,” MRS Proceedings. 2009. link Times cited: 0 Abstract: Understanding thermal transport between carbon nanotubes (CN… read moreAbstract: Understanding thermal transport between carbon nanotubes (CNTs) and dielectric substrates is important both for nanoscale thermal management and CNT device applications. We investi-gate thermal transport between a (10,10) CNT and an SiO2 substrate through non-equilibrium classical molecular dynamics (MD) simulations. The thermal boundary conductance (TBC) is computed by setting up a temperature pulse in the CNT and monitoring its relaxation. The TBC is found to scale nearly linearly with temperature between 200�600 K, where a quantum correction is applied to the CNT heat capacity through its phonon density of states. However, the TBC ap-pears most sensitive to the strength the CNT-substrate interaction, which linearly modulates it between 0.05�0.30 WK- 1 m- 1 , in the range suggested by recent experimental data. read less USED (low confidence) W. Malfait, “Short- and medium-range order in silicate glasses and melts: insights from Raman and NMR spectroscopy and effects on bulk melt properties.” 2007. link Times cited: 0 NOT USED (low confidence) H. Song, H. Dong, W. Dong, and Y. Luo, “Atomic-Level Insights into Hollow Silica-Based Materials for Drug Delivery: Effects of Wettability and Porosity.,” ACS biomaterials science & engineering. 2023. link Times cited: 0 Abstract: Experimental evidence has demonstrated that the drug carrier… read moreAbstract: Experimental evidence has demonstrated that the drug carrier capacity can be significantly enhanced through the use of hollow silica particles. Nevertheless, the effects of varying functional drug carrier surfaces and porous structures remain ambiguous. This study employs molecular dynamics simulations to examine the effects of varying the surface wettability, pore size, and flow velocity on the transfer process. The different levels of wettability of the silica surface with the coarse-grained water model is illustrated by adjusted interaction parameters. The effect of wettability is investigated. With weak interactions, the flow molecules form a nanodroplet to transfer through the porous structure. A strong interaction will lead to molecules flowing as a liquid film to transfer through the structure. Interestingly, the "contradiction effect" is observed when the flow molecules fail to penetrate the porous structure with weak interactions, during which surface tension dominates their flow behavior. Moreover, different porous structures are considered. The flow behaviors are divided into three processes: (1) fast flowing, (2) transient point, and (3) penetration flowing. Furthermore, the concept of surface molecules is defined to quantitatively measure the effect of porosity. A recommended contact angle is proposed. The results will pave the way for more carrier structures in medical engineering. read less NOT USED (low confidence) G. Zhao et al., “Understanding the role of engineered cluster evolution in enhancing Ag layer growth on oxides,” Applied Surface Science. 2023. link Times cited: 0 NOT USED (low confidence) S. Z. Pakzad et al., “Nanomechanical Modeling of the Bending Response of Silicon Nanowires,” ACS Applied Nano Materials. 2023. link Times cited: 4 NOT USED (low confidence) M. Owen et al., “Modelling the impact of configurational entropy on the stability of amorphous SiO2,” Scripta Materialia. 2023. link Times cited: 0 NOT USED (low confidence) C. Ugwumadu et al., “Structure, vibrations and electronic transport in silicon suboxides: Application to physical unclonable functions,” Journal of Non-Crystalline Solids: X. 2023. link Times cited: 1 NOT USED (low confidence) T. Hsu et al., “Score-based denoising for atomic structure identification,” ArXiv. 2022. link Times cited: 2 Abstract: We propose an effective method for removing thermal vibratio… read moreAbstract: We propose an effective method for removing thermal vibrations that complicate the task of analyzing complex dynamics in atomistic simulation of condensed matter. Our method iteratively subtracts thermal noises or perturbations in atomic positions using a denoising score function trained on synthetically noised but otherwise perfect crystal lattices. The resulting denoised structures clearly reveal underlying crystal order while retaining disorder associated with crystal defects. Purely geometric, agnostic to interatomic potentials, and trained without inputs from explicit simulations, our denoiser can be applied to simulation data generated from vastly different interatomic interactions. The denoiser is shown to improve existing classification methods such as common neighbor analysis and polyhedral template matching, reaching perfect classification accuracy on a recent benchmark dataset of thermally perturbed structures up to the melting point. Demonstrated here in a wide variety of atomistic simulation contexts, the denoiser is general, robust, and readily extendable to delineate order from disorder in structurally and chemically complex materials. read less NOT USED (low confidence) J. Liu, P. Buahom, C. Lu, H. Yu, and C. B. Park, “Microscopic revelation of the solid–gas coupling and Knudsen effect on the thermal conductivity of silica aerogel with inter-connected pores,” Scientific Reports. 2022. link Times cited: 2 NOT USED (low confidence) S. Haseen and P. Kroll, “Analyzing the Effect of Composition, Density, and the Morphology of the ‘free’ Carbon Phase on Elastic Moduli in Silicon Oxycarbide Ceramics,” Journal of the European Ceramic Society. 2022. link Times cited: 1 NOT USED (low confidence) S. Z. Pakzad, M. N. Esfahani, and B. E. Alaca, “The Role of Native Oxide on the Mechanical Behavior of Silicon Nanowires,” SSRN Electronic Journal. 2022. link Times cited: 5 NOT USED (low confidence) M. Yang, Q. Sheng, L. Guo, H. Zhang, and G. Tang, “How Gas-Solid Interaction Matters in Graphene-Doped Silica Aerogels.,” Langmuir : the ACS journal of surfaces and colloids. 2022. link Times cited: 5 Abstract: It was interesting to experimentally find that the thermal i… read moreAbstract: It was interesting to experimentally find that the thermal insulation of silica aerogels was improved by doping graphene sheets with high heat conductivity. The underlying mechanism is investigated in the present work from the perspective of gas-solid interaction using a comprehensive analysis of molecular dynamics (MD) simulations, theoretical modeling, and experimental data. The MD-modeled small pores are demonstrated to effectively represent big pores in silica aerogels because of similar heat conduction physics, because it is found that adsorption does not contribute to gas heat conduction. Meanwhile, based on the experimentally measured density, the porous structures are schematically re-engineered using molecular modeling for the first time. The evaluated pore size distributions numerically present a consistency with available experimental data. Inspired by the visualization of the 3D pore structure, we proposed a graphene/silica/nitrogen model to evaluate the role of graphene in heat conduction: it can not only reduce effective gas collision (impede heat transport) but also enhance the gas-solid coupling effect. The former is dominant because of the high porosity, leading to an improvement in thermal insulation. The competition between them can be the reason for the "trade-off" phenomenon in the graphene doping effect in the available experiment. read less NOT USED (low confidence) D. Martí, E. Martín-Martinez, J. Torras, O. Betran, P. Turon, and C. Aléman, “In silico study of substrate chemistry effect on the tethering of engineered antibodies for SARS-CoV-2 detection: Amorphous silica vs gold,” Colloids and Surfaces. B, Biointerfaces. 2022. link Times cited: 1 NOT USED (low confidence) I. Camacho et al., “On the anticorrosion mechanism of molten salts based nanofluids,” Solar Energy Materials and Solar Cells. 2022. link Times cited: 5 NOT USED (low confidence) C. Hou et al., “Atomistic Simulation toward Real-scale Microprocessor Circuits,” Chemical Physics Letters. 2022. link Times cited: 1 NOT USED (low confidence) M. Eghbalian, R. Ansari, and S. Rouhi, “Effects of geometrical parameters and functionalization percentage on the mechanical properties of oxygenated single-walled carbon nanotubes,” Journal of Molecular Modeling. 2021. link Times cited: 8 NOT USED (low confidence) N. Eydi, S. Feghhi, and H. Jafari, “Comprehensive approach to determination of space proton-induced displacement defects in silica optical fiber,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2021. link Times cited: 4 NOT USED (low confidence) J. Luo, Y. Cheng, C. Zhou, T. Sinno, and L. Liu, “A general approach for calculating melt–solid impurity segregation coefficients based on thermodynamic integration,” Journal of Applied Physics. 2021. link Times cited: 1 Abstract: The equilibrium segregation of impurities at the melt–solid … read moreAbstract: The equilibrium segregation of impurities at the melt–solid interface during silicon crystallization is a key factor in determining the impurity concentration and distribution in the crystal. Unfortunately, this property is difficult to measure experimentally due to the presence of complex transport physics in the melt. Here, using the Tersoff family of empirical potential models, we describe a thermodynamic integration framework for computing the interstitial oxygen and substitutional carbon segregation coefficients in silicon. Thermodynamic integration using an ideal gas reference state for the impurity atoms is shown to be an efficient and convenient pathway for evaluating impurity chemical potentials in both solid and liquid phases. We find that the segregation coefficient is captured well for substitutional carbon impurity while it is significantly underestimated for interstitial oxygen. The latter discrepancy is partially attributed to the qualitatively incorrect silicon solid-to-liquid density ratio predicted by the empirical interatomic potential. read less NOT USED (low confidence) H. Ghasemi, H. Yazdani, E. Fini, and Y. Mansourpanah, “Interactions of SARS-CoV-2 with inanimate surfaces in built and transportation environments,” Sustainable Cities and Society. 2021. link Times cited: 6 NOT USED (low confidence) D. A. Husseini et al., “Surface Functionalization Utilizing Mesoporous Silica Nanoparticles for Enhanced Evanescent-Field Mid-Infrared Waveguide Gas Sensing,” Coatings. 2021. link Times cited: 8 Abstract: This work focuses on the development of nanoparticle-based l… read moreAbstract: This work focuses on the development of nanoparticle-based layer-by-layer (LbL) coatings for enhancing the detection sensitivity and selectivity of volatile organic compounds (VOCs) using on-chip mid-infrared (MIR) waveguides (WGs). First, we demonstrate construction of conformal coatings of polymer/mesoporous silica nanoparticles (MSNs) on the surface of Si-based WGs using the LbL technique and evaluate the coating deposition conditions, such as pH and substrate withdrawal speed, on the thickness and homogeneity of the assemblies. We then use the modified WGs to achieve enhanced sensitivity and selectivity of polar organic compounds, such as ethanol, versus non-polar ones, such as methane, in the MIR region. In addition, using density functional theory calculations, we show that such an improvement in sensing performance is achieved due to preferential adsorption of ethanol molecules within MSNs in the vicinity of the WG evanescent field. read less NOT USED (low confidence) A. Rohskopf, S. Wyant, K. Gordiz, H. R. Seyf, M. G. Muraleedharan, and A. Henry, “Fast & accurate interatomic potentials for describing thermal vibrations,” Computational Materials Science. 2020. link Times cited: 7 NOT USED (low confidence) S. Kiyohara, M. Tsubaki, and T. Mizoguchi, “Learning excited states from ground states by using an artificial neural network,” npj Computational Materials. 2020. link Times cited: 13 NOT USED (low confidence) J. J. G. Moreno, K. Pan, Y. Wang, and W. Li, “A computational study of APTES surface functionalization of diatom-like amorphous SiO2 surfaces for heavy metal adsorption.,” Langmuir : the ACS journal of surfaces and colloids. 2020. link Times cited: 18 Abstract: The amorphous silica (SiO2) shell on diatom frustules is a h… read moreAbstract: The amorphous silica (SiO2) shell on diatom frustules is a highly attractive biomaterial for removing pollutants from aquatic ecosystems. The surface activity of silica can be enhanced by modification with organosilanes. In this work, we present an atomic-level theoretical study based on Molecular Dynamics (MD) and dispersion-corrected Density Functional Theory (DFT-D3BJ) calculations on the surface stability and adsorption of heavy metal compounds on silane and APTES covered SiO2 surfaces. Our simulations show that at low APTES coverage, molecular adsorption of Cd(OH)2 and HgCl2 is more favourable near the modifier, compared to As(OH)3 that binds at the hydroxylated region on silica. At higher coverages, the metallic compounds are preferentially adsorbed by the terminating amino group on the surface, whereas the adsorption in the region between APTES and the oxide surface is also spontaneous. The adsorption is strongly driven by van der Waals interactions at the highly-covered surface, where the consideration of dispersion corrections reduces the modifier-adsorbate interatomic distances and increases the adsorption energy by c.a. 0.4-0.7 eV. The adsorption of water is favourable, although it is generally weaker than for the heavy metal compounds. Based on our results, we conclude that the addition of APTES modifiers on silica increases the adsorption strength and provides extra binding sites for the adsorption of heavy metal pollutants. These outcomes can be used for the design of more efficient biomaterials' structures for heavy metals depollution. read less NOT USED (low confidence) T. Loeffler, T. Patra, H. Chan, M. Cherukara, and S. Sankaranarayanan, “Active Learning the Potential Energy Landscape for Water Clusters from Sparse Training Data,” Journal of Physical Chemistry C. 2020. link Times cited: 19 Abstract: Molecular dynamics with predefined functional forms is a pop… read moreAbstract: Molecular dynamics with predefined functional forms is a popular technique for understanding dynamical evolution of systems. The predefined functional forms impose limits on the physics that can be... read less NOT USED (low confidence) E. Okulich, V. Okulich, and D. Tetelbaum, “Impact of Oxygen Vacancies on the Formation and Structure of Filaments in SiO2-Based Memristors,” Technical Physics Letters. 2020. link Times cited: 4 NOT USED (low confidence) S. Takamoto, S. Izumi, and J. Li, “TeaNet: universal neural network interatomic potential inspired by iterative electronic relaxations,” ArXiv. 2019. link Times cited: 29 NOT USED (low confidence) Z. Liu, T. Yunqing, N. Liao, and P. Yang, “Study on interfacial interaction between Si and ZnO,” Ceramics International. 2019. link Times cited: 15 NOT USED (low confidence) N. Nayir, A. V. van Duin, and S. Erkoç, “Development of a ReaxFF Reactive Force Field for Interstitial Oxygen in Germanium and Its Application to GeO2/Ge Interfaces,” The Journal of Physical Chemistry C. 2019. link Times cited: 10 Abstract: We developed the ReaxFF force field parameters for Ge/O/H in… read moreAbstract: We developed the ReaxFF force field parameters for Ge/O/H interactions, specifically targeted for the applications of Ge/GeO2 interfaces and O-diffusion in bulk Ge. The original training set, taken... read less NOT USED (low confidence) D. Fang et al., “Spider-Web-Inspired Nanocomposite-Modified Separator: Structural and Chemical Cooperativity Inhibiting the Shuttle Effect in Li-S Batteries.,” ACS nano. 2019. link Times cited: 70 Abstract: Despite their high theoretical capacity density (1675 mAh g-… read moreAbstract: Despite their high theoretical capacity density (1675 mAh g-1), the application of Li-S batteries has been seriously hindered by the shuttle effect of polysulfides. Here, inspired by the working principle of natural spider webs, we synthesized a spider-web-like nanocomposite in which many hollow mesoporous silica (mSiO2) nanospheres/Co nanoparticles were threaded by interconnected nitrogen-doped carbon nanotubes (NCNTs). Then the nanocomposite (denoted as Co/mSiO2-NCNTs) was coated on the commercial separator by a simple infiltration to mitigate the above issue. The intimate combination of three-dimensional conductive networks (NCNTs) with abundant polysulfide adsorbent sites (SiO2 and N)/polysulfide conversion catalysts (Co and Co-N x species) allows the Co/mSiO2-NCNTs coating layer to not only effectively capture polysulfides via both physical confinement and chemical bonding but also accelerate the redox kinetics of polysulfides significantly. Furthermore, the combination of ex situ experiment and theoretical calculation demonstrates that the reversible adsorption/desorption of polysulfides on mSiO2 nanospheres benefits uniform deposition of Li2S2/Li2S on the conductive networks, which contributes to long-term cycling stability. As a result, Li-S batteries with Co/mSiO2-NCNTs-coated separators exhibited both excellent cycling stability and rate performance. read less NOT USED (low confidence) F. Meng, J. Liu, and R. Richards, “Effect of water vapor on the thermal resistance between amorphous silica nanoparticles,” Journal of Applied Physics. 2018. link Times cited: 4 Abstract: Nanoparticle-based materials are of interest because of thei… read moreAbstract: Nanoparticle-based materials are of interest because of their unique thermal properties. Possessing the lowest thermal conductivities of any solid materials known, they have been widely used as insulating materials. However, the presence of water vapor has been shown to have a large influence on those properties. In this work, we investigate the effect of water vapor on the heat transfer between nanoparticles using non-equilibrium molecular dynamics simulations. We calculate the absolute thermal resistance and Kapitza resistance between adjacent amorphous spherical silica nanoparticles, when water molecules are allowed to diffuse as vapor into the interstitial pores between particles. The thermal resistance between nanoparticles is shown to decrease rapidly when water vapor is introduced into the pores between particles. The largest decrease in interparticle resistance occurs as a result of the silanization of the silica particle surfaces. A secondary decrease is attributable to the liquid bridge that forms as water molecules condense around the contact point between nanoparticles. Most of the decrease in resistance between nanoparticles occurs when water vapor is first introduced at relative humidities (rh) of less than 1%. As the relative humidity increases above 1%, the interparticle thermal resistance decreases more slowly, approaching a constant value near 50% rh. Numerical results are compared to experimental measurements of heat transfer across packed beds of 20 nm silica nanoparticles exposed to water vapor. The simulation results are shown to be consistent with the experimental measurements for relative humidities below 15% rh, while underpredicting the experimental measurements above 15% rh.Nanoparticle-based materials are of interest because of their unique thermal properties. Possessing the lowest thermal conductivities of any solid materials known, they have been widely used as insulating materials. However, the presence of water vapor has been shown to have a large influence on those properties. In this work, we investigate the effect of water vapor on the heat transfer between nanoparticles using non-equilibrium molecular dynamics simulations. We calculate the absolute thermal resistance and Kapitza resistance between adjacent amorphous spherical silica nanoparticles, when water molecules are allowed to diffuse as vapor into the interstitial pores between particles. The thermal resistance between nanoparticles is shown to decrease rapidly when water vapor is introduced into the pores between particles. The largest decrease in interparticle resistance occurs as a result of the silanization of the silica particle surfaces. A secondary decrease is attributable to the liquid bridge that for... read less NOT USED (low confidence) H. Liu et al., “Confinement Impact for the Dynamics of Supported Metal Nanocatalyst.,” Small. 2018. link Times cited: 5 Abstract: Supported metal nanoparticles play key roles in nanoelectron… read moreAbstract: Supported metal nanoparticles play key roles in nanoelectronics, sensors, energy storage/conversion, and catalysts for the sustainable production of fuels and chemicals. Direct observation of the dynamic processes of nanocatalysts at high temperatures and the confinement of supports is of great significance to investigate nanoparticle structure and functions for practical utilization. Here, in situ high-resolution transmission electron microscopy photos and videos are combined with dynamics simulations to reveal the real-time dynamic behavior of Pt nanocatalysts at operation temperatures. Amorphous Pt surface on moving and deforming particles is the working structure during the high operation temperature rather than a static crystal surface and immobilization on supports as proposed before. The free rearrangement of the shape of Pt nanoparticles allows them to pass through narrow windows, which is generally considered to immobilize the particles. The Pt particles, no matter what their sizes, prefer to stay inside nanopores even when they are fast moving near an opening at temperatures up to 900 °C. The porous confinement also blocks the sintering of the particles under the confinement size of pores. These contribute to the continuous high activity and stability of Pt nanocatalysts inside nanoporous supports during a long-term evaluation of catalytic reforming reaction. read less NOT USED (low confidence) X. Zhao et al., “Breaking the Current‐Retention Dilemma in Cation‐Based Resistive Switching Devices Utilizing Graphene with Controlled Defects,” Advanced Materials. 2018. link Times cited: 179 Abstract: Cation‐based resistive switching (RS) devices, dominated by … read moreAbstract: Cation‐based resistive switching (RS) devices, dominated by conductive filaments (CF) formation/dissolution, are widely considered for the ultrahigh density nonvolatile memory application. However, the current‐retention dilemma that the CF stability deteriorates greatly with decreasing compliance current makes it hard to decrease operating current for memory application and increase driving current for selector application. By centralizing/decentralizing the CF distribution, this current‐retention dilemma of cation‐based RS devices is broken for the first time. Utilizing the graphene impermeability, the cation injecting path to the RS layer can be well modulated by structure‐defective graphene, leading to control of the CF quantity and size. By graphene defect engineering, a low operating current (≈1 µA) memory and a high driving current (≈1 mA) selector are successfully realized in the same material system. Based on systematically materials analysis, the diameter of CF, modulated by graphene defect size, is the major factor for CF stability. Breakthrough in addressing the current‐retention dilemma will instruct the future implementation of high‐density 3D integration of RS memory immune to crosstalk issues. read less NOT USED (low confidence) M. Dewapriya and R. Rajapakse, “Development of a homogenous nonlinear spring model characterizing the interfacial adhesion properties of graphene with surface defects,” Composites Part B-engineering. 2016. link Times cited: 16 NOT USED (low confidence) M. Dilamian and A. Haghi, “Update on Aerogels Material and Technology.” 2015. link Times cited: 0 NOT USED (low confidence) M. Dilamian, “Understanding Modeling and Simulation of Aerogels Behavior: From Theory to Application.” 2015. link Times cited: 0 NOT USED (low confidence) A. Sogoyan and V. A. Polunin, “A model for the formation of leakage currents in the dielectrics of MOS structures under the effect of heavy charged particles,” Russian Microelectronics. 2015. link Times cited: 6 NOT USED (low confidence) A. Sogoyan and V. A. Polunin, “A model for the formation of leakage currents in the dielectrics of MOS structures under the effect of heavy charged particles,” Russian Microelectronics. 2015. link Times cited: 0 NOT USED (low confidence) Y. Ni, Y. Chalopin, and S. Volz, “Few layer graphene based superlattices as efficient thermal insulators,” Applied Physics Letters. 2013. link Times cited: 18 Abstract: While graphene and few layer graphene (FLG) are considered a… read moreAbstract: While graphene and few layer graphene (FLG) are considered as having the highest thermal conductivity in their in-plane directions, our molecular dynamics (MD) simulations however show that those systems are also characterized by a superior thermal contact resistance, which could be largely tuned with the layer number when in contact with a silica substrate. Taking advantages of such a resistive interface, MD simulations show that SiO2/FLG superlattices have a thermal conductivity as low as 0.30 W/m K, exhibiting a promising prospect in nano-scale thermal insulation. These findings pave the way for an improved thermal management of nanoscale systems such as thermal barrier coatings and phase change memory materials with atomic-scale super-insulators. read less NOT USED (low confidence) J. Lowther, “The Role Played by Computation in Understanding Hard Materials,” Materials. 2011. link Times cited: 8 Abstract: In the last decade, computation has played a valuable role i… read moreAbstract: In the last decade, computation has played a valuable role in the understanding of materials. Hard materials, in particular, are only part of the application. Although materials involving B, C, N or O remain the most valued atomic component of hard materials, with diamond retaining its distinct superiority as the hardest, other materials involving a wide variety of metals are proving important. In the present work the importance of both ab-initio approaches and molecular dynamics aspects will be discussed with application to quite different systems. On one hand, ab-initio methods are applied to lightweight systems and advanced nitrides. Following, the use of molecular dynamics will be considered with application to strong metals that are used for high temperature applications. read less NOT USED (low confidence) R. Jones, C. Weinberger, S. Coleman, and G. Tucker, “Introduction to Atomistic Simulation Methods.” 2016. link Times cited: 1 NOT USED (low confidence) T. Ng, S. Joshi, J. Yeo, and Z. Liu, “Effects of Nanoporosity on the Mechanical Properties and Applications of Aerogels in Composite Structures.” 2016. link Times cited: 2 NOT USED (low confidence) A. Galashev, “The Spectral Properties of (SiO 2 ) n , (GaN) m , (GaAs) m , (SiO 2 ) n (GaN) m , and (SiO 2 ) n (GaAs) m Nanoparticles: Computer Experiment.” 2016. link Times cited: 0 NOT USED (low confidence) A. Galashev, “The Spectral Properties of (SiO2) n , (GaN) m , (GaAs) m , (SiO2) n (GaN) m , and (SiO2) n (GaAs) m Nanoparticles: Computer Experiment.” 2015. link Times cited: 0 NOT USED (low confidence) S. Tyaginov et al., “Impact of the Surrounding Network on the Si-O Bond-Breakage Energetics,” MRS Proceedings. 2009. link Times cited: 0 Abstract: We extend the McPherson Model for silicon-oxygen bond-breaka… read moreAbstract: We extend the McPherson Model for silicon-oxygen bond-breakage derived for a single SiO4 tetrahedron to capture the influence of the whole lattice. Several pair-wise potentials have been compared in the model including Mie-Gruneisen as well as diverse forms of TTAM/BKS. The contribution of the whole lattice substantially increases the activation energy for the Si-O bond rupture. The corresponding small transition rate of a non-distorted Si-O bond suggests that the interaction with the electric field alone can not be responsible for the bond-breakage and the contribution of other components such as energy delivered by particles and/or bond weakening is required. read less NOT USED (high confidence) A. Koneru et al., “Multi-reward reinforcement learning based development of inter-atomic potential models for silica,” npj Computational Materials. 2023. link Times cited: 0 NOT USED (high confidence) Y. Sun et al., “Determining the interlayer shearing in twisted bilayer MoS2 by nanoindentation,” Nature Communications. 2022. link Times cited: 11 NOT USED (high confidence) L. C. Erhard, J. Rohrer, K. Albe, and V. L. Deringer, “A machine-learned interatomic potential for silica and its relation to empirical models,” npj Computational Materials. 2022. link Times cited: 32 NOT USED (high confidence) F. Font-Clos et al., “Predicting the failure of two-dimensional silica glasses,” Nature Communications. 2022. link Times cited: 14 NOT USED (high confidence) Z. Tian et al., “Atomistic Insights into Aluminum Doping Effect on Surface Roughness of Deposited Ultra-Thin Silver Films,” Nanomaterials. 2021. link Times cited: 6 Abstract: Ultra-thin and continuous metallic silver films are attracti… read moreAbstract: Ultra-thin and continuous metallic silver films are attracting growing interest due to the applications in flexible transparent conducting electrodes. The surface morphology and structure of silver film are very important for its electrical resistivity and optical loss. Therefore, roughness control is essential for the production of ultra-thin metallic electrode film. We have investigated the effect of aluminum doping on the improvement of surface morphology of ultra-thin silver films using molecular dynamics simulations. Al-doped silver films showed smaller surface roughness than pure silver films at various substrate temperatures. When the temperature of the substrate was 600 K, the roughness of Al-doped silver film first decreased, and then increased with the increase of the incident velocity of silver atoms. Silver atoms were more likely to agglomerate on the surface of the substrate after adding aluminum atoms, as aluminum dopants promoted the immobilization of silver atoms on SiO2 substrate due to the anchoring effect. The smoother surface could be attributable to the reduced mean free path of silver due to the cage effect by the aluminum dopant. read less NOT USED (high confidence) C. Qian and J. Wang, “The effects of stacking mode and thickness on the frictional behaviour of multilayer silicene,” RSC Advances. 2020. link Times cited: 0 Abstract: Understanding the contact behaviour of 2D materials in nanos… read moreAbstract: Understanding the contact behaviour of 2D materials in nanoscale is of great importance for their applications. In the present work, molecular dynamics simulation is employed to study the frictional behaviour of the AA′ and AB stacked multilayer silicene for up to 4 layers placed on the weakly adhesive amorphous SiO2 substrate with a sliding AFM tip. During the sliding process, the AFM cantilever represented by virtual atoms moves with the velocity of 2 m s−1 along the zigzag direction under a load of 2 nN at 300 K. The stick-slip frictional behaviour shows high sensitivity to the number of layers. As the thickness increases, the friction force first increases from the monolayer to bilayer and then decreases from the bilayer to 4-layer, which shows an exotic tendency for the first time among all the reported lamellar materials to date where the friction usually decreases monotonically with thickness. For all the investigated thicknesses, the friction on AA′-stacked silicene is slightly larger than the AB stacked counterpart, and the difference diminishes with increasing thickness. The frictional behaviour of AA′ bilayer presents the highest peak force with evolving weakening phenomenon induced by a phase transition to the planar structure. Herein, we analyze the frictional force distribution on the tip with kurtosis and skewness as measurement parameters for the commensurability and rigidity components, respectively. The contact area between silicene and the diamond tip is compared for different silicene morphologies. The result shows an affinity between friction and rigidity of multilayer silicene, which is closely related to the interlayer covalent bonds and limited shear between sublayers. read less NOT USED (high confidence) W. Xu, Y. Jiao, and J. Fish, “An atomistically-informed multiplicative hyper-elasto-plasticity-damage model for high-pressure induced densification of silica glass,” Computational Mechanics. 2020. link Times cited: 0 NOT USED (high confidence) W. Xu, Y. Jiao, and J. Fish, “An atomistically-informed multiplicative hyper-elasto-plasticity-damage model for high-pressure induced densification of silica glass,” Computational Mechanics. 2020. link Times cited: 6 NOT USED (high confidence) G. Chen, J. Chen, and Z.-liang Wang, “Thermal Transport at Interface Between Single-Layer Graphene and Water Film,” International Journal of Thermophysics. 2020. link Times cited: 2 NOT USED (high confidence) S. Damián-Vázquez, J. F. Alvarado, and E. O. Castrejón-González, “A combined force field for the silica/nickel system,” Molecular Simulation. 2020. link Times cited: 2 Abstract: ABSTRACT Silica and nickel are frequently used in the synthe… read moreAbstract: ABSTRACT Silica and nickel are frequently used in the synthesis of carbon nanotubes by the chemical vapour deposition (CVD) process. The molecular simulation of this process requires the knowledge of force fields to model the interactions occurring between those species at the atomic scale. This work proposes a combined force field to model the silica/nickel system when these components are in contact as substrate and catalyst, respectively, in the CVD process. The proposed combined force field includes the Lennard–Jones (n–m) potential for modelling the silicon/nickel pair interactions and the Buckingham potential for the oxygen/nickel pair interactions. The combined force field is completed by the Tersoff potential to model silica (SiO2) and the Sutton–Chen potential for the cohesive forces present in the nickel clusters. Parameters for the Lennard–Jones (n–m) and Buckingham pair potentials were fitted, by the least squares technique, to interaction energies data for the silica/nickel system. The energies were obtained from Ab-initio (DFT) calculations using the VASP code. It was found that the combined force field reproduces adequately, by molecular dynamics simulation, the adherence (adsorption) of nickel clusters on the silica surface. Keeping stable this configuration is crucial in modelling the carbon nanotubes synthesis by the CVD process. read less NOT USED (high confidence) M. Vorholzer, J. Vilhena, R. Pérez, E. Gnecco, D. Dietzel, and A. Schirmeisen, “Temperature Activates Contact Aging in Silica Nanocontacts,” Physical Review X. 2019. link Times cited: 7 Abstract: Matthias Vorholzer, J. G. Vilhena, Ruben Perez, Enrico Gnecc… read moreAbstract: Matthias Vorholzer, J. G. Vilhena, Ruben Perez, Enrico Gnecco, Dirk Dietzel, and André Schirmeisen Institute of Applied Physics, Justus-Liebig-Universität Giessen, 35392 Giessen, Germany Department of Physics, University of Basel, Klingelbergstrasse 82, 4056 Basel, Switzerland Departamento de Física Teórica de la Materia Condensada, Universidad Autónoma de Madrid, E-28049 Madrid, Spain Condensed Matter Physics Center (IFIMAC), Universidad Autónoma de Madrid, E-28049 Madrid, Spain Otto Schott Institute of Materials Research, Friedrich Schiller University Jena, 07742 Jena, Germany Center for Materials Research, Justus-Liebig-Universität Giessen, 35392 Giessen, Germany read less NOT USED (high confidence) Z. Fan, Y. Wang, X. Gu, P. Qian, Y. Su, and T. Ala‐Nissila, “A minimal Tersoff potential for diamond silicon with improved descriptions of elastic and phonon transport properties,” Journal of Physics: Condensed Matter. 2019. link Times cited: 10 Abstract: Silicon is an important material and many empirical interato… read moreAbstract: Silicon is an important material and many empirical interatomic potentials have been developed for atomistic simulations of it. Among them, the Tersoff potential and its variants are the most popular ones. However, all the existing Tersoff-like potentials fail to reproduce the experimentally measured thermal conductivity of diamond silicon. Here we propose a modified Tersoff potential and develop an efficient open source code called GPUGA (graphics processing units genetic algorithm) based on the genetic algorithm and use it to fit the potential parameters against energy, virial and force data from quantum density functional theory calculations. This potential, which is implemented in the efficient open source GPUMD (graphics processing units molecular dynamics) code, gives significantly improved descriptions of the thermal conductivity and phonon dispersion of diamond silicon as compared to previous Tersoff potentials and at the same time well reproduces the elastic constants. Furthermore, we find that quantum effects on the thermal conductivity of diamond silicon at room temperature are non-negligible but small: using classical statistics underestimates the thermal conductivity by about 10% as compared to using quantum statistics. read less NOT USED (high confidence) A. Dmitriev, A. Nikonov, W. Österle, and B. Jim, “VERIFICATION OF RABINOWICZ’ CRITERION BY DIRECT MOLECULAR DYNAMICS MODELING,” Facta Universitatis, Series: Mechanical Engineering. 2019. link Times cited: 6 Abstract: In the paper we use direct molecular dynamics modeling to va… read moreAbstract: In the paper we use direct molecular dynamics modeling to validate the criterion for formation of wear debris proposed by E. Rabinowicz in 1958. A conventional molecular dynamics using a classical Tersoff’s potential was applied to simulate the sliding behavior within a thin film corresponding to a tribofilm formed from silica nano-particles in amorphous-like state. The simulation was carried out by varying the initial temperature and the spatial size of the simulated crystallite. The results show the change in sliding behavior of silica-based tribofilm depending on the temperature and the size parameter of the system under consideration. Thus increasing the temperature provides smooth sliding while at moderate conditions wear process can occur via debris formation. Our estimations show good correlation between predicted critical size of the simulated system and calculated energetic characteristics. read less NOT USED (high confidence) D. Melgar, M. Lauricella, G. O’Brien, and N. J. English, “Amplitude effects on seismic velocities: How low can we go?,” The Journal of chemical physics. 2019. link Times cited: 1 Abstract: α-quartz is one of the most important SiO2 polymorphs becaus… read moreAbstract: α-quartz is one of the most important SiO2 polymorphs because it is the basis of very common minerals, especially for seabed materials with geoscientific importance. The elastic characterization of these materials is particularly relevant when the properties governing phonon and sound propagation are involved. These studies are especially interesting for oil exploration purposes. Recently, we published a new method that constitutes to the best of our knowledge the first attempt to recreate longitudinal and transversal perturbations in a simulation box to observe their propagation through the crystal by means of a set of descriptors [D. Melgar et al., J. Phys. Chem. C 122, 3006-3013 (2018)]. The agreement with the experimental S- and P-wave velocities was rather excellent. Thus, an effort has been undertaken to deepen the particularities of this new methodology. Here, bearing in mind this encouraging initial methodology-development progress, we deepen our knowledge of the particularities of this new methodology in presenting a systematic investigation of the implementation of the perturbation source. This includes new ways of creating the perturbation, as well as analyzing the possible effects the perturbation amplitude could have on the resultant velocities. In addition, different force fields were tested to describe the interatomic interactions. The lack of dependence of the seismic velocities on the way the perturbation is created and the perturbation amplitude, and the good agreement with the experimental results are the main reasons that allow the definition of this new methodology as robust and reliable. These qualities are consolidated by the physical behavior of the calculated velocities in the presence of vacancies and under stress. The development of this method opens up a new line of research of calculating seismic velocities for geophysically relevant materials in a systematic way, with full control not only on the sample features (composition, porosity, vacancies, stress, etc.) but also on the particularities of perturbation itself, as well as determining optimal system-response metrics. read less NOT USED (high confidence) Y. Lysogorskiy, T. Hammerschmidt, J. Janssen, J. Neugebauer, and R. Drautz, “Transferability of interatomic potentials for molybdenum and silicon,” Modelling and Simulation in Materials Science and Engineering. 2019. link Times cited: 14 Abstract: Interatomic potentials are widely used in computational mate… read moreAbstract: Interatomic potentials are widely used in computational materials science, in particular for simulations that are too computationally expensive for density functional theory (DFT). Most interatomic potentials have a limited application range and often there is very limited information available regarding their performance for specific simulations. We carried out high-throughput calculations for molybdenum and silicon with DFT and a number of interatomic potentials. We compare the DFT reference calculations and experimental data to the predictions of the interatomic potentials. We focus on a large number of basic materials properties, including the cohesive energy, atomic volume, elastic coefficients, vibrational properties, thermodynamic properties, surface energies and vacancy formation energies, which enables a detailed discussion of the performance of the different potentials. We further analyze correlations between properties as obtained from DFT calculations and how interatomic potentials reproduce these correlations, and suggest a general measure for quantifying the accuracy and transferability of an interatomic potential. From our analysis we do not establish a clearcut ranking of the potentials as each potential has its strengths and weaknesses. It is therefore essential to assess the properties of a potential carefully before application of the potential in a specific simulation. The data presented here will be useful for selecting a potential for simulations of Mo or Si. read less NOT USED (high confidence) S. Suryavanshi, A. Gabourie, A. B. Farimani, and E. Pop, “Thermal boundary conductance of two-dimensional MoS2 interfaces,” Journal of Applied Physics. 2019. link Times cited: 29 Abstract: Understanding the thermal properties of two-dimensional (2D)… read moreAbstract: Understanding the thermal properties of two-dimensional (2D) materials and devices is essential for thermal management of 2D applications. Here we perform molecular dynamics simulations to evaluate both the specific heat of $MoS_{2}$ as well as the thermal boundary conductance (TBC) between one to five layers of $MoS_{2}$ with amorphous $SiO_{2}$ and between single-layer $MoS_{2}$ and crystalline $AlN$. The results of all calculations are compared to existing experimental data. In general, the TBC of such 2D interfaces is low, below ~20 $MWm^{-2}K^{-1}$, due to the weak van der Waals (vdW) coupling and mismatch of phonon density of states (PDOS) between materials. However, the TBC increases with vdW coupling strength, with temperature, and with the number of $MoS_{2}$ layers (which introduce additional phonon modes). These findings suggest that the TBC of 2D materials is tunable by modulating their interface interaction, the number of layers, and finding a PDOS-matched substrate, with important implications for future energy-efficient 2D electronics, photonics, and thermoelectrics. read less NOT USED (high confidence) J. Chen et al., “Mechanical properties and deformation behaviors of surface-modified silicon: a molecular dynamics study,” Journal of Materials Science. 2018. link Times cited: 16 NOT USED (high confidence) K. Termentzidis et al., “Enhanced thermal conductivity in percolating nanocomposites: a molecular dynamics investigation.,” Nanoscale. 2018. link Times cited: 6 Abstract: In this work we present a molecular dynamics investigation o… read moreAbstract: In this work we present a molecular dynamics investigation of thermal transport in a silica-gallium nitride nanocomposite. A surprising enhancement of the thermal conductivity for crystalline volume fractions larger than 5% is found, which cannot be predicted by an effective medium approach, not even including percolation effects, the model systematically leading to an underestimation of the effective thermal conductivity. The behavior can instead be reproduced if an effective volume fraction twice larger than the real one is assumed, which translates into a percolation effect surprisingly stronger than the usual one. Such a scenario can be understood in terms of a phonon tunneling between inclusions, enhanced by the iso-orientation of all particles. Indeed, if a misorientation is introduced, the thermal conductivity strongly decreases. We also show that a percolating nanocomposite clearly stands in a different position than other nanocomposites, where thermal transport is dominated by the interface scattering and where parameters such as the interface density play a major role, differently from our case. read less NOT USED (high confidence) Y. Xu, F. Zhu, M. Wang, X. Liu, and S. Liu, “Molecular Dynamics Simulation on Grinding Process of Cu-Si and Cu-SiO2 Composite Structures,” 2018 19th International Conference on Electronic Packaging Technology (ICEPT). 2018. link Times cited: 2 Abstract: The molecular dynamics (MD) simulation was performed for Cu-… read moreAbstract: The molecular dynamics (MD) simulation was performed for Cu-Si and Cu-SiO2 nano-metric grinding models. The grinding depth and speed were considered to investigate influence of them in grinding. The transformation of the atomic crystal structure of the workpiece during the grinding process was investigated to reveal the material removal mechanism in nano-grinding. The variation of grinding force between the two models was analyzed. The results showed that grinding force was mainly composed of tangential force and normal force. Under the same grinding parameters, the grinding force of the two models changed similarly. read less NOT USED (high confidence) A. Dmitriev, A. Nikonov, and W. Österle, “Molecular Dynamics Modeling of the Sliding Performance of an Amorphous Silica Nano-Layer—The Impact of Chosen Interatomic Potentials,” Lubricants. 2018. link Times cited: 7 Abstract: The sliding behavior of an amorphous silica sample between t… read moreAbstract: The sliding behavior of an amorphous silica sample between two rigid surfaces is in the focus of the present paper. Molecular Dynamics using a classical Tersoff’s potential and a recently developed ReaxFF potential was applied for simulating sliding within a thin film corresponding to a tribofilm formed from silica nanoparticles. The simulations were performed at different temperatures corresponding to moderate and severe tribological stressing conditions. Simulations with both potentials revealed the need of considering different temperatures in order to obtain a sound interpretation of experimental findings. The results show the striking differences between the two potentials not only in terms of magnitude of the resistance stress (about one order of magnitude) but also in terms of friction mechanisms. The expected smooth sliding regime under high temperature conditions was predicted by both simulations, although with Tersoff’s potential smooth sliding was obtained only at the highest temperature. On the other hand, at room temperature Tersoff-style calculations demonstrate stick-slip behavior, which corresponds qualitatively with our experimental findings. Nevertheless, comparison with a macroscopic coefficient of friction is not possible because simulated resistance stresses do not depend on the applied normal pressure. read less NOT USED (high confidence) J. Chen et al., “Nanoindentation and deformation behaviors of silicon covered with amorphous SiO2: a molecular dynamic study,” RSC Advances. 2018. link Times cited: 26 Abstract: A fundamental understanding of the mechanical properties and… read moreAbstract: A fundamental understanding of the mechanical properties and deformation behaviors of surface modified silicon during chemical mechanical polishing (CMP) processes is difficult to obtain at the nanometer scale. In this research, MD simulations of monocrystalline silicon covered with an amorphous SiO2 film with different thickness are implemented by nanoindentation, and it is found that both the indentation modulus and hardness increase with the growing indentation depth owning to the strongly silicon substrate effect. At the same indentation depth, the indentation modulus decreases shapely with the increase of film thickness because of less substrate influence, while the hardness agrees well with the trend of modulus at shallow depth but mismatches at larger indentation depth. The observed SiO2 film deformation consists of densification and thinning along indentation direction and extension in the deformed area due to the rotation and deformation of massive SiO4 tetrahedra. The SiO2 film plays an important role in the onset and development of silicon phase transformation. The thinner the SiO2 film is, the earlier the silicon phase transformation takes place. So the numbers of phase transformation atoms increase with the decrease of SiO2 film thickness at the same indentation depth. It is suggested that the thicker film should be better during CMP process for higher material removal rate and less defects within silicon substrate. read less NOT USED (high confidence) J. Rimsza, R. Jones, and L. Criscenti, “Crack propagation in silica from reactive classical molecular dynamics simulations,” Journal of the American Ceramic Society. 2018. link Times cited: 32 NOT USED (high confidence) F. Grigoriev, V. Sulimov, and A. Tikhonravov, “Simulation of the optical coating deposition,” Advanced Optical Technologies. 2018. link Times cited: 5 Abstract: A brief review of the mathematical methods of thin-film grow… read moreAbstract: A brief review of the mathematical methods of thin-film growth simulation and results of their applications is presented. Both full-atomistic and multi-scale approaches that were used in the studies of thin-film deposition are considered. The results of the structural parameter simulation including density profiles, roughness, porosity, point defect concentration, and others are discussed. The application of the quantum level methods to the simulation of the thin-film electronic and optical properties is considered. Special attention is paid to the simulation of the silicon dioxide thin films. read less NOT USED (high confidence) M. Verdier et al., “Influence of amorphous layers on the thermal conductivity of phononic crystals,” Physical Review B. 2018. link Times cited: 16 Abstract: The impact of amorphous phases around the holes and at the u… read moreAbstract: The impact of amorphous phases around the holes and at the upper and lower free surfaces on thermal transport in silicon phononic membranes is studied. By means of molecular dynamics and Monte Carlo simulations, we explore the impact of the amorphous phase (oxidation and amorphous silicon), surfaces roughness, and a series of geometric parameters on thermal transport. We show that the crystalline phase drives the phenomena; the two main parameters are (i) the crystalline fraction between two holes and (ii) the crystalline thickness of the membranes. We reveal the hierarchical impact of nanostructurations on the thermal conductivity, namely, from the most resistive to the less resistive: the creation of holes, the amorphous phase around them, and the amorphization of the membranes edges. The surfaces or interfaces perpendicular to the heat flow hinder the thermal conductivity to a much greater extent than those parallel to the heat flow. read less NOT USED (high confidence) Z. Ong, B. Qiu, S. Xu, X. Ruan, and E. Pop, “Flexural resonance mechanism of thermal transport across graphene-SiO2 interfaces,” Journal of Applied Physics. 2018. link Times cited: 24 Abstract: Understanding the microscopic mechanism of heat dissipation … read moreAbstract: Understanding the microscopic mechanism of heat dissipation at the dimensionally mismatched interface between a two-dimensional (2D) crystal and its substrate is crucial for the thermal management of devices based on 2D materials. Here, we study the lattice contribution to thermal (Kapitza) transport at graphene-SiO2 interfaces using molecular dynamics (MD) simulations and non-equilibrium Green's functions (NEGF). We find that 78 percent of the Kapitza conductance is due to sub-20 THz flexural acoustic modes, and that a resonance mechanism dominates the interfacial phonon transport. MD and NEGF estimate the classical Kapitza conductance to be hK ≈ 10 to 16 MW K−1 m−2 at 300 K, respectively, consistent with existing experimental observations. Taking into account quantum mechanical corrections, this value is approximately 28% lower at 300 K. Our calculations also suggest that hK scales as T2 at low temperatures (T < 100 K) due to the linear frequency dependence of phonon transmission across the graphene-SiO2 interface at low frequencies. Our study sheds light on the role of flexural acoustic phonons in heat dissipation from graphene to its substrate.Understanding the microscopic mechanism of heat dissipation at the dimensionally mismatched interface between a two-dimensional (2D) crystal and its substrate is crucial for the thermal management of devices based on 2D materials. Here, we study the lattice contribution to thermal (Kapitza) transport at graphene-SiO2 interfaces using molecular dynamics (MD) simulations and non-equilibrium Green's functions (NEGF). We find that 78 percent of the Kapitza conductance is due to sub-20 THz flexural acoustic modes, and that a resonance mechanism dominates the interfacial phonon transport. MD and NEGF estimate the classical Kapitza conductance to be hK ≈ 10 to 16 MW K−1 m−2 at 300 K, respectively, consistent with existing experimental observations. Taking into account quantum mechanical corrections, this value is approximately 28% lower at 300 K. Our calculations also suggest that hK scales as T2 at low temperatures (T < 100 K) due to the linear frequency dependence of phonon transmission across the graphene-SiO... read less NOT USED (high confidence) H. Seyf, W.-L. Lv, A. Rohskopf, and A. Henry, “The Importance of Phonons with Negative Phase Quotient in Disordered Solids,” Scientific Reports. 2018. link Times cited: 6 NOT USED (high confidence) R. Ranganathan, Y. Shi, and P. Keblinski, “Commonalities in frequency-dependent viscoelastic damping in glasses in the MHz to THz regime,” Journal of Applied Physics. 2017. link Times cited: 10 Abstract: We use non-equilibrium molecular dynamics oscillatory shear … read moreAbstract: We use non-equilibrium molecular dynamics oscillatory shear simulations to study frequency-dependent viscoelastic damping spanning nearly six decades in frequency range (MHz to THz), in a wide range of model glasses including binary glasses such as Cu-Zr metallic glass (MG), Wahnstrom glass and amorphous silica, and unary glasses, namely, Dzugutov glass and amorphous silicon. First, for the Cu-Zr MG, we elucidate the role of quench rate, number of shear cycles, shear amplitude, and shear temperature on the damping characteristics. We observe striking commonalities in damping characteristics for all glasses studied—(i) a peak in the loss modulus in the high-frequency regime (∼THz) and (ii) persistent damping in the low-frequency regime (extending down to 10 s of MHz). The high-frequency peak is seen to overlap with the range of natural vibrational frequencies for each glass, and arises from coupling between the excited harmonic vibrational modes. On the other hand, persistent damping at intermediate and lo... read less NOT USED (high confidence) A. Stepanov and D. Tetelbaum, “Molecular dynamics simulation of the penetration of silicon by hypersonic waves generated in native silicon oxide under irradiation,” Journal of Surface Investigation: X-ray, Synchrotron and Neutron Techniques. 2017. link Times cited: 3 NOT USED (high confidence) M. L. Nietiadi et al., “The bouncing threshold in silica nanograin collisions.,” Physical chemistry chemical physics : PCCP. 2017. link Times cited: 17 Abstract: Using molecular dynamics simulations, we study collisions be… read moreAbstract: Using molecular dynamics simulations, we study collisions between amorphous silica nanoparticles. Our silica model contains uncontaminated surfaces, that is, the effect of surface hydroxylation or of adsorbed water layers is excluded. For central collisions, we characterize the boundary between sticking and bouncing collisions as a function of impact velocity and particle size and quantify the coefficient of restitution. We show that the traditional Johnson-Kendall-Roberts (JKR) model provides a valid description of the ingoing trajectory of two grains up to the moment of maximum compression. The distance of closest approach is slightly underestimated by the JKR model, due to the appearance of plasticity in the grains, which shows up in the form of localized shear transformation zones. The JKR model strongly underestimates the contact radius and the collision duration during the outgoing trajectory, evidencing that the breaking of covalent bonds during grain separation is not well described by this model. The adhesive neck formed between the two grains finally collapses while creating narrow filaments joining the grains, which eventually tear. read less NOT USED (high confidence) B. Cowen and M. El-Genk, “Estimates of point defect production in α-quartz using molecular dynamics simulations,” Modelling and Simulation in Materials Science and Engineering. 2017. link Times cited: 5 Abstract: Molecular dynamics (MD) simulations are performed to investi… read moreAbstract: Molecular dynamics (MD) simulations are performed to investigate the production of point defects in α-quartz by oxygen and silicon primary knock-on atoms (PKAs) of 0.25–2 keV. The Wigner–Seitz (WS) defect analysis is used to identify the produced vacancies, interstitials, and antisites, and the coordination defect analysis is used to identify the under and over-coordinated oxygen and silicon atoms. The defects at the end of the ballistic phase and the residual defects, after annealing, increase with increased PKA energy, and are statistically the same for the oxygen and silicon PKAs. The WS defect analysis results show that the numbers of the oxygen vacancies and interstitials (VO, Oi) at the end of the ballistic phase is the highest, followed closely by those of the silicon vacancies and interstitials (VSi, Sii). The number of the residual oxygen and silicon vacancies and interstitials are statistically the same. In addition, the under-coordinated OI and SiIII, which are the primary defects during the ballistic phase, have high annealing efficiencies (>89%). The over-coordinated defects of OIII and SiV, which are not nearly as abundant in the ballistic phase, have much lower annealing efficiencies (<63%) that decrease with increased PKA energy. read less NOT USED (high confidence) J. Kioseoglou, M. Katsikini, K. Termentzidis, I. K. Karakostas, and E. Paloura, “Mechanism and crucial parameters on GaN nanocluster formation in a silica matrix,” Journal of Applied Physics. 2017. link Times cited: 7 Abstract: The formation of wurtzite GaN nanoclusters in an amorphous s… read moreAbstract: The formation of wurtzite GaN nanoclusters in an amorphous silica matrix, via gallium and nitrogen ion implantation and rapid thermal annealing, is identified using Extended X Ray Absorption Fine Structure analysis. The mechanism and the crucial parameters that rule the formation of the nanoclusters are established by the use of molecular dynamics simulations. The dominant structural parameters are found to be the concentration of the silicon and oxygen vacancies that are formed during the implantation and the annealing temperature. It is concluded that annealing at 1400 K and 8% Ga/Si and 12% N/O ratios are needed for the formation of GaN nanoclusters. In addition to that, the GaN nanocluster formation is accomplished only when the vacancy concentrations of silicon and oxygen atoms are equal to 10% and 20%, respectively. Finally, the observation of various snapshots upon an increase of the annealing duration indicates the coalescence of smaller GaN nuclei towards larger ones, designating that the Ostwald... read less NOT USED (high confidence) Y. Qi, J. Liu, J. Zhang, Y. Dong, and Q. Li, “Wear Resistance Limited by Step Edge Failure: The Rise and Fall of Graphene as an Atomically Thin Lubricating Material.,” ACS applied materials & interfaces. 2017. link Times cited: 67 Abstract: Owing to its intrinsically lubricious property, graphene has… read moreAbstract: Owing to its intrinsically lubricious property, graphene has a high potential to be an atomically thin solid lubricant for sliding interfaces. Despite its ultrahigh breaking strength at the nanoscale, graphene often fails to maintain its integrity when subjected to macroscale tribological tests. To reveal the true wear characteristics of graphene, a nanoscale diamond tip was used to scratch monolayer graphene mechanically exfoliated to SiO2 substrates. Our experimental results show that while graphene exhibited extraordinary wear resistance in the interior region, it could be easily damaged at the step edge under a much lower normal load (∼2 orders of magnitude smaller). Similar behavior with substantially reduced wear resistance at the edge was also observed for monatomic graphene layer on graphite surface. Using molecular dynamics simulations, we attributed this markedly weak wear resistance at the step edge to two primary mechanisms, i.e., atom-by-atom adhesive wear and peel induced rupture. Our findings shed light on the paradox that graphene is nanoscopically strong yet macroscopically weak. As step edge is ubiquitous for two-dimensional materials at the macroscale, our study also provides a guiding direction for maximizing the mechanical and tribological performance of these atomically thin materials. read less NOT USED (high confidence) W. Shou and H. Pan, “Silicon-wall interfacial free energy via thermodynamics integration.,” The Journal of chemical physics. 2016. link Times cited: 7 Abstract: We compute the interfacial free energy of a silicon system i… read moreAbstract: We compute the interfacial free energy of a silicon system in contact with flat and structured walls by molecular dynamics simulation. The thermodynamics integration method, previously applied to Lennard-Jones potentials [R. Benjamin and J. Horbach, J. Chem. Phys. 137, 044707 (2012)], has been extended and implemented in Tersoff potentials with two-body and three-body interactions taken into consideration. The thermodynamic integration scheme includes two steps. In the first step, the bulk Tersoff system is reversibly transformed to a state where it interacts with a structureless flat wall, and in a second step, the flat structureless wall is reversibly transformed into an atomistic SiO2 wall. Interfacial energies for liquid silicon-wall interfaces and crystal silicon-wall interfaces have been calculated. The calculated interfacial energies have been employed to predict the nucleation mechanisms in a slab of liquid silicon confined by two walls and compared with MD simulation results. read less NOT USED (high confidence) S. Takamoto et al., “Charge-transfer interatomic potential for investigation of the thermal-oxidation growth process of silicon,” Journal of Applied Physics. 2016. link Times cited: 11 Abstract: A charge-transfer interatomic potential, based on the hybrid… read moreAbstract: A charge-transfer interatomic potential, based on the hybrid-Tersoff potential that incorporates a covalent-ionic mixed-bond nature, was developed to reproduce the growth process of the thermal oxidation of silicon. A fitting process was employed with various reference structures sampled by MD. Actively exploring and learning the wide-range of phase space enabled us to develop a robust interatomic potential. Our interatomic potential reproduced the bulk properties of Si and SiO2 polymorphs well, in addition to the radial distribution function and bond angle distribution of amorphous SiO2. The covalent-ionic mixed-bond nature of the interatomic potential well reproduced the dissociation process of an oxygen molecule on the Si/SiO2 interface. The initial oxidation simulation was performed on the silicon surface. We grew the amorphous SiO2 layer by incorporating the oxygen molecules into the silicon network at the interface. The density of the SiO2 layer and the charge distribution at the interface showed go... read less NOT USED (high confidence) S. Chowdhury, B. Haque, and J. Gillespie, “Molecular dynamics simulations of the structure and mechanical properties of silica glass using ReaxFF,” Journal of Materials Science. 2016. link Times cited: 92 NOT USED (high confidence) J. L. Braun et al., “Size effects on the thermal conductivity of amorphous silicon thin films,” Physical Review B. 2016. link Times cited: 87 Abstract: In this study, we investigate thickness-limited size effects… read moreAbstract: In this study, we investigate thickness-limited size effects on the thermal conductivity of amorphous silicon thin films ranging from 3 to 1636 nm grown via sputter deposition. While exhibiting a constant value up to ~100 nm, the thermal conductivity increases with film thickness thereafter. The thickness dependence we demonstrate is ascribed to boundary scattering of long wavelength vibrations and an interplay between the energy transfer associated with propagating modes (propagons) and nonpropagating modes (diffusons). A crossover from propagon to diffuson modes is deduced to occur at a frequency of ~1.8 THz via simple analytical arguments. These results provide empirical evidence of size effects on the thermal conductivity of amorphous silicon and systematic experimental insight into the nature of vibrational thermal transport in amorphous solids. read less NOT USED (high confidence) A. France-Lanord, P. Soukiassian, C. Glattli, and E. Wimmer, “Ab initio parameterization of a charge optimized many-body forcefield for Si-SiO2: Validation and thermal transport in nanostructures.,” The Journal of chemical physics. 2016. link Times cited: 13 Abstract: In an effort to extend the reach of current ab initio calcul… read moreAbstract: In an effort to extend the reach of current ab initio calculations to simulations requiring millions of configurations for complex systems such as heterostructures, we have parameterized the third-generation Charge Optimized Many-Body (COMB3) potential using solely ab initio total energies, forces, and stress tensors as input. The quality and the predictive power of the new forcefield are assessed by computing properties including the cohesive energy and density of SiO2 polymorphs, surface energies of alpha-quartz, and phonon densities of states of crystalline and amorphous phases of SiO2. Comparison with data from experiments, ab initio calculations, and molecular dynamics simulations using published forcefields including BKS (van Beest, Kramer, and van Santen), ReaxFF, and COMB2 demonstrates an overall improvement of the new parameterization. The computed temperature dependence of the thermal conductivity of crystalline alpha-quartz and the Kapitza resistance of the interface between crystalline Si(001) and amorphous silica is in excellent agreement with experiment, setting the stage for simulations of complex nanoscale heterostructures. read less NOT USED (high confidence) A. M. Tamidi and Y. Sasajima, “The Relationship between Nanocluster Precipitation and Thermal Conductivity in Si/Ge Amorphous Multilayer Films,” Journal of Nanomaterials. 2016. link Times cited: 2 Abstract: We have used a molecular dynamics technique to simulate the … read moreAbstract: We have used a molecular dynamics technique to simulate the relationship between nanocluster precipitation and thermal conductivity in Si/Ge amorphous multilayer films, with and without Cu addition. In the study, the Green-Kubo equation was used to calculate thermal conductivity in these materials. Five specimens were prepared: Si/Ge layers, Si/Ge + Cu layers, Si + Cu/Ge + Cu layers, Si/Cu/Ge/Cu layers, and Si/Cu/Ge layers. The number of precipitated nanoclusters in these specimens, which is defined as the number of four-coordinate atoms, was counted along the lateral direction of the specimens. The observed results of precipitate formation were considered in relation to the thermal conductivity results. Enhancement of precipitation of nanoclusters by Cu addition, that is, densification of four-coordinate atoms, can prevent the increment of thermal conductivity. Cu dopant increases the thermal conductivity of these materials. Combining these two points, we concluded that Si/Cu/Ge is the best structure to improve the conversion efficiency of the Si/Ge amorphous multilayer films. read less NOT USED (high confidence) C. Y. Chuang, S. Han, L. Zepeda-Ruiz, and T. Sinno, “On coarse projective integration for atomic deposition in amorphous systems.,” The Journal of chemical physics. 2015. link Times cited: 2 Abstract: Direct molecular dynamics simulation of atomic deposition un… read moreAbstract: Direct molecular dynamics simulation of atomic deposition under realistic conditions is notoriously challenging because of the wide range of time scales that must be captured. Numerous simulation approaches have been proposed to address the problem, often requiring a compromise between model fidelity, algorithmic complexity, and computational efficiency. Coarse projective integration, an example application of the "equation-free" framework, offers an attractive balance between these constraints. Here, periodically applied, short atomistic simulations are employed to compute time derivatives of slowly evolving coarse variables that are then used to numerically integrate differential equations over relatively large time intervals. A key obstacle to the application of this technique in realistic settings is the "lifting" operation in which a valid atomistic configuration is recreated from knowledge of the coarse variables. Using Ge deposition on amorphous SiO2 substrates as an example application, we present a scheme for lifting realistic atomistic configurations comprised of collections of Ge islands on amorphous SiO2 using only a few measures of the island size distribution. The approach is shown to provide accurate initial configurations to restart molecular dynamics simulations at arbitrary points in time, enabling the application of coarse projective integration for this morphologically complex system. read less NOT USED (high confidence) P. Brault and E. Neyts, “Molecular dynamics simulations of supported metal nanocatalyst formation by plasma sputtering,” Catalysis Today. 2015. link Times cited: 26 NOT USED (high confidence) Z. Jing, L. Xin, and H. Sun, “Replica exchange reactive molecular dynamics simulations of initial reactions in zeolite synthesis.,” Physical chemistry chemical physics : PCCP. 2015. link Times cited: 11 Abstract: Molecular simulation is a promising tool for the study of ze… read moreAbstract: Molecular simulation is a promising tool for the study of zeolite formation. However, sufficient sampling remains a grand challenge for the practical use of molecular simulation for this purpose. Here, we investigate the initial stage of zeolite synthesis under realistic conditions by using the replica-exchange method and the ReaxFF reactive force field. After a total simulation time of 480 ns, both energetic and structural properties approach convergence. Analyses of data collected at 600 K show that the inorganic structure directing agent NaOH promotes the aggregation of silicate, the formation of branched Si atoms and the formation of 5-membered rings. With the trajectories collected simultaneously at different temperatures, the effect of temperature is discussed. read less NOT USED (high confidence) J. Wang, A. Rajendran, and A. Dongare, “Atomic scale modeling of shock response of fused silica and α-quartz,” Journal of Materials Science. 2015. link Times cited: 34 NOT USED (high confidence) P. Käshammer and T. Sinno, “A mechanistic study of impurity segregation at silicon grain boundaries,” Journal of Applied Physics. 2015. link Times cited: 28 Abstract: The segregation behavior of carbon and oxygen atoms at vario… read moreAbstract: The segregation behavior of carbon and oxygen atoms at various silicon grain boundaries was studied using a combination of atomistic simulation and analytical modeling. First, quasi-lattice Grand Canonical Monte Carlo simulations were used to compute segregation isotherms as a function of grain boundary type, impurity atom loading level, and temperature. Next, the atomistic results were employed to regress different analytical segregation models and extract thermodynamic and structural properties. The multilayer Brunauer–Emmett–Teller (BET) isotherm was found to quantitatively capture all the simulation conditions probed in this work, while simpler, single layer models such as the Langmuir-McLean model did not. Some of the BET parameters, namely, the binding free energy of the first adsorption layer and the impurity holding capacity of each layer, were tested for correlation with various measures of grain boundary structure and/or mechanical properties. It was found that certain measures of the atomistic stress distribution correlate strongly with the first-layer binding free energy for substitutional carbon atoms, while common grain boundary identifiers such as sigma value and energy density are not useful in this regard. Preliminary analysis of the more complex case of interstitial oxygen segregation showed that similar measures based on atomistic stress also may be useful here, but more systematic correlative studies are needed to develop a comprehensive picture. read less NOT USED (high confidence) W.-L. Lv and A. Henry, “Non-negligible Contributions to Thermal Conductivity From Localized Modes in Amorphous Silicon Dioxide,” Scientific Reports. 2015. link Times cited: 58 NOT USED (high confidence) W.-L. Lv and A. Henry, “Direct calculation of modal contributions to thermal conductivity via Green–Kubo modal analysis,” New Journal of Physics. 2015. link Times cited: 115 Abstract: We derived a new method for direct calculation of the modal … read moreAbstract: We derived a new method for direct calculation of the modal contributions to thermal conductivity, which is termed Green–Kubo modal analysis (GKMA). The GKMA method combines the lattice dynamics formalism with the Green–Kubo formula for thermal conductivity, such that the thermal conductivity becomes a direct summation of modal contributions, where one need not define the phonon velocity. As a result, the GKMA method can be applied to any material/group of atoms, where the atoms vibrate around stable equilibrium positions, which includes non-stoichiometric compounds, random alloys, amorphous materials and even rigid molecules. By using molecular dynamics simulations to obtain the time history of each mode’s contribution to the heat current, one naturally includes anharmonicity to full order and can obtain insight into the interactions between different modes through the cross-correlations. As an example, we applied the GMKA method to crystalline and amorphous silicon. The modal contributions at each frequency result from the analysis and thereby allow one to apply a quantum correction to the mode heat capacity to determine the temperature dependence of thermal conductivity. The predicted temperature dependent thermal conductivity for amorphous silicon shows the best agreement with experiments to date. The GKMA method provides new insight into the nature of phonon transport, as it casts the problem in terms of mode–mode correlation instead of scattering, and provides a general unified formalism that can be used to understand phonon–phonon interactions in essentially any class of materials or structures where the atoms vibrate around stable equilibrium sites. read less NOT USED (high confidence) S. Lee and W. Lu, “Controlling the number of graphene sheets exfoliated from graphite by designed normal loading and frictional motion,” Journal of Applied Physics. 2014. link Times cited: 8 Abstract: We use molecular dynamics to study the exfoliation of patter… read moreAbstract: We use molecular dynamics to study the exfoliation of patterned nanometer-sized graphite under various normal loading conditions for friction-induced exfoliation. Using highly ordered pyrolytic graphite (HOPG) as well as both amorphous and crystalline SiO2 substrate as example systems, we show that the exfoliation process is attributed to the corrugation of the HOPG surface and the atomistic roughness of the substrate when they contact under normal loading. The critical normal strain, at which the exfoliation occurs, is higher on a crystalline substrate than on an amorphous substrate. This effect is related to the atomistic flatness and stiffness of the crystalline surface. We observe that an increase of the van der Waals interaction between the graphite and the substrate results in a decrease of the critical normal strain for exfoliation. We find that the magnitude of the normal strain can effectively control the number of exfoliated graphene layers. This mechanism suggests a promising approach of applyi... read less NOT USED (high confidence) B. Deng, A. Chernatynskiy, M. Khafizov, D. Hurley, and S. Phillpot, “Kapitza resistance of Si/SiO2 interface,” Journal of Applied Physics. 2014. link Times cited: 61 Abstract: A phonon wave packet dynamics method is used to characterize… read moreAbstract: A phonon wave packet dynamics method is used to characterize the Kapitza resistance of a Si/SiO2 interface in a Si/SiO2/Si heterostructure. By varying the thickness of SiO2 layer sandwiched between two Si layers, we determine the Kapitza resistance for the Si/SiO2 interface from both wave packet dynamics and a direct, non-equilibrium molecular dynamics approach. The good agreement between the two methods indicates that they have each captured the anharmonic phonon scatterings at the interface. Moreover, detailed analysis provides insights as to how individual phonon mode scatters at the interface and their contribution to the Kapitza resistance. read less NOT USED (high confidence) J. Lei, Z. Liu, J. Yeo, and T. Ng, “Determination of the Young’s modulus of silica aerogels – an analytical–numerical approach,” Soft Matter. 2013. link Times cited: 33 Abstract: The superior thermal, optical, acoustical, and mechanical pr… read moreAbstract: The superior thermal, optical, acoustical, and mechanical properties of a nano-porous and ultra-light material called silica aerogel have been known to science since the 1930's. In this study, we propose a new analytical–numerical model of a silica aerogel to predict its Young's modulus. The molecular dynamics (MD) simulation method was adopted to model and simulate the backbone of silica aerogels with different densities in an improved negative pressure rupturing method, and their mechanical properties were investigated. To generate the model using MD, the traditional method was improved by changing the sequence of its procedures, and it was proven to be a more stable and physically representative method. In the prediction, we proposed a two-level nano-porous structure model according to our simulations and the widely accepted fractal structure of silica aerogels. The Young's modulus of a silica aerogel, which is shown in a power–law relationship with the density of samples, was derived by the two-level hierarchical model and uniaxial tension tests. We envisage that this new model can be applied in more analytical–numerical studies to reveal other interesting mechanical properties of silica aerogels. read less NOT USED (high confidence) J. Yeo, Z. S. Liu, and T. Ng, “Enhanced thermal characterization of silica aerogels through molecular dynamics simulation,” Modelling and Simulation in Materials Science and Engineering. 2013. link Times cited: 29 Abstract: Porous structures of silica aerogels are generated using cla… read moreAbstract: Porous structures of silica aerogels are generated using classical molecular dynamics, with the Tersoff potential, which has been re-parametrized for modeling silicon dioxides. This work demonstrates that this potential is superior to the widely used BKS potential in terms of characterizing the thermal conductivities of amorphous silica, by comparing the vibrational density of states with previous experimental studies. Aerogel samples of increasing densities are obtained through an expanding, heating and quenching process. Reverse non-equilibrium molecular dynamics is applied at each density to determine the thermal conductivity. A power-law fit of the results is found to accurately reflect the power-law variation found in experimental bulk aerogels. The results are also of the same order of magnitude as experimental bulk aerogels, but they are consistently higher. By analyzing the pore size distribution on different simulation length scales, we show that such a disparity is due to finite sizes of pores that can be represented, where increasing simulation length scales lead to an increase in the largest pore size that can be modeled. read less NOT USED (high confidence) A. Galashev and O. Rakhmanova, “Temperature changes of the optical properties of (SiO2)n, (GaAs)m, and (SiO2)n(GaAs)m nanoparticles: Computer experiment,” High Temperature. 2013. link Times cited: 6 NOT USED (high confidence) J. Zhang, C. Liu, Y. Shu, and J. Fan, “Growth and properties of Cu thin film deposited on Si(0 0 1) substrate: A molecular dynamics simulation study,” Applied Surface Science. 2012. link Times cited: 50 NOT USED (high confidence) X. Zhang, M. Hu, K. Giapis, and D. Poulikakos, “Schemes for and Mechanisms of Reduction in Thermal Conductivity in Nanostructured Thermoelectrics,” Journal of Heat Transfer-transactions of The Asme. 2012. link Times cited: 20 Abstract: Nonequilibrium molecular dynamics (NEMD) simulations were pe… read moreAbstract: Nonequilibrium molecular dynamics (NEMD) simulations were performed to investigate schemes for enhancing the energy conversion efficiency of thermoelectric nanowires (NWs), including (1) roughening of the nanowire surface, (2) creating nanoparticle inclusions in the nanowires, and (3) coating the nanowire surface with other materials. The enhancement in energy conversion efficiency was inferred from the reduction in thermal conductivity of the nanowire, which was calculated by imposing a temperature gradient in the longitudinal direction. Compared to pristine nanowires, our simulation results show that the schemes proposed above lead to nanocomposite structures with considerably lower thermal conductivity (up to 82% reduction), implying ~5X enhancement in the ZT coefficient. This significant effect appears to have two origins: (1) increase in phonon-boundary scattering and (2) onset of interfacial interference. The results suggest new fundamental–yet realizable ways to improve markedly the energy conversion efficiency of nanostructured thermoelectrics. read less NOT USED (high confidence) P. T. Araujo et al., “In situ atomic force microscopy tip-induced deformations and Raman spectroscopy characterization of single-wall carbon nanotubes.,” Nano letters. 2012. link Times cited: 16 Abstract: In this work, an atomic force microscope (AFM) is combined w… read moreAbstract: In this work, an atomic force microscope (AFM) is combined with a confocal Raman spectroscopy setup to follow in situ the evolution of the G-band feature of isolated single-wall carbon nanotubes (SWNTs) under transverse deformation. The SWNTs are pressed by a gold AFM tip against the substrate where they are sitting. From eight deformed SWNTs, five exhibit an overall decrease in the Raman signal intensity, while three exhibit vibrational changes related to the circumferential symmetry breaking. Our results reveal chirality dependent effects, which are averaged out in SWNT bundle measurements, including a previously elusive mode symmetry breaking that is here explored using molecular dynamics calculations. read less NOT USED (high confidence) S. Zhao, J. Xue, Y. Wang, and S. Yan, “Effect of SiO2 substrate on the irradiation-assisted manipulation of supported graphene: a molecular dynamics study,” Nanotechnology. 2012. link Times cited: 49 Abstract: The irradiation effects in graphene supported by SiO2 substr… read moreAbstract: The irradiation effects in graphene supported by SiO2 substrate including defect production and implantation efficiency are investigated using the molecular dynamics (MD) method with empirical potentials. We show that the irradiation damage in supported graphene comes from two aspects: the direct damage induced by the incident ions and the indirect damage resulting from backscattered particles and sputtered atoms from the substrate. In contrast with the damage in suspended graphene, we find that the indirect damage is dominant in supported graphene at high energies. As a result, enhanced irradiation damage in supported graphene is observed when the incident energy is above 5 keV for Ar and 3 keV for Si. The direct damage probability at all energies, even the total damage probability at low energies, in supported graphene is always much lower than that in suspended graphene because of the higher threshold displacement energy of carbon atoms. In addition, we demonstrate the striking finding that it is possible to dope graphene with sputtered atoms from the substrate and the implantation probability is considerable at optimal energies. Our results indicate that the substrate is an important factor in the process of ion-irradiation-assisted engineering of the properties of graphene. read less NOT USED (high confidence) T. E. Letsoalo and J. Lowther, “Elastic and thermodynamic properties of potentially superhard carbon boride materials,” Journal of Superhard Materials. 2012. link Times cited: 6 NOT USED (high confidence) S. Tyaginov, V. Sverdlov, I. Starkov, W. Gös, and T. Grasser, “Impact of O-Si-O bond angle fluctuations on the Si-O bond-breakage rate,” Microelectron. Reliab. 2009. link Times cited: 2 NOT USED (high confidence) S. Tyaginov, V. Sverdlov, W. Gos, and T. Grasser, “Statistics of Si-O Bond-Breakage Rate Variations Induced by O-Si-O Angle Fluctuations,” 2009 13th International Workshop on Computational Electronics. 2009. link Times cited: 1 Abstract: The McPherson model for the Si-O bond-breakage has been exte… read moreAbstract: The McPherson model for the Si-O bond-breakage has been extended in a manner to capture the effect of O-Si-O angle variations on the breakage rate. Using a distribution function of the O-Si-O bond angle, a series of breakage rate probability densities has been calculated as a function of the applied electric field. Using such a distribution function we have calculated the mean vale and the standard deviation of the breakage rate and compare them to the nominal rate corresponding to the fixed angle of 109.48deg observed in crystalline alpha-quartz. It is shown that the mean value of this rate is substantially higher than and the standard deviation is comparable with the nominal rate. Obtained dependencies demonstrate a linear trend in a log-fin space, thereby validating the thermo-chemical model for the time-dependent-dielectric breakdown also in the case of non-uniform O-Si-O angle distribution typical for amorphous silica. read less NOT USED (high confidence) Z. Xue-Yang, C. Jun, and H. Wang-Yu, “Atomic simulation of surface damage in fused silica under laser irradiation,” Acta Physica Sinica. 2023. link Times cited: 0 Abstract: Fused silica optical element is the core component of the in… read moreAbstract: Fused silica optical element is the core component of the inertial confinement nuclear fusion ignition device. Due to the demanding ignition conditions of the device for high power laser, the damage of fused silica optical element under strong laser is the key to restrict the operation of the ignition device. Therefore, the study of the surface damage of fused silica irradiated by laser is crucial to the development of the ignition device for inertial confinement nuclear fusion. In this paper, large-scale non-equilibrium molecular dynamics simulation method and micro-structure analysis technology suitable for dynamic process are proposed to study the damage process of fused silica surface under laser loading. Based on the theoretical study of high-temperature plasma fireball model, the damage of high-temperature fused silica plasma ball to surface is simulated. By tracking the local structure, temperature distribution and surface morphology, the factors affecting the surface damage of fused silica are analyzed. Our researches show that the size, distance from the surface, and temperature of high-temperature fused silica balls have important effects on the surface damage. We find that there are two different damage modes under the combined effect of the above factors. One is with a rapid damage process, generating U-shaped voids and no further obvious damages after the surface spraying, and the other is with a slow damage process, continuous expansion and resulting in a larger damage area. The surface morphologies formed by these two damage modes is consistent with the two typical damage morphologies observed in the experiments. This article can provide a guidance for understanding the complex damage process in fused silica under laser irradiation. read less NOT USED (high confidence) D. Meng et al., “Gamma-ray irradiation-induced oxidation and disproportionation at the amorphous SiO2/Si interfaces,” Journal of Materials Chemistry C. 2020. link Times cited: 2 Abstract: Distinct interfacial structure changes, including oxidation … read moreAbstract: Distinct interfacial structure changes, including oxidation and disproportionation, have been found to be the main response to the Mrad dose gamma ray irradiation for SiO2/Si films. read less NOT USED (high confidence) S. Urata and S. Li, “Higher order Cauchy–Born rule based multiscale cohesive zone model and prediction of fracture toughness of silicon thin films,” International Journal of Fracture. 2016. link Times cited: 18 NOT USED (high confidence) J. Yeo, “Modeling and simulation of the structural evolution and thermal properties of ultralight aerogel and 2D materials.” 2014. link Times cited: 1 |
 Tersoff_LAMMPS_MunetohMotookaMoriguchi_2007_SiO__MO_501246546792_000
Tersoff_LAMMPS_MunetohMotookaMoriguchi_2007_SiO__MO_501246546792_000