Citations
This panel presents information regarding the papers that have cited the interatomic potential (IP) whose page you are on.
The OpenKIM machine learning based Deep Citation framework is used to determine whether the citing article actually used the IP in computations (denoted by "USED") or only provides it as a background citation (denoted by "NOT USED"). For more details on Deep Citation and how to work with this panel, click the documentation link at the top of the panel.
The word cloud to the right is generated from the abstracts of IP principle source(s) (given below in "How to Cite") and the citing articles that were determined to have used the IP in order to provide users with a quick sense of the types of physical phenomena to which this IP is applied.
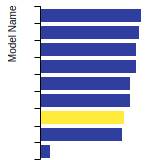
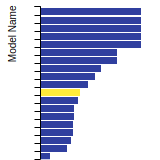
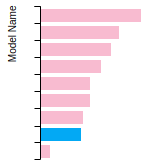
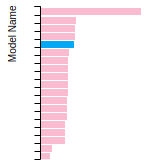
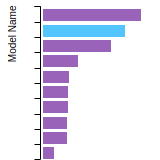
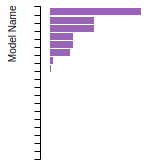
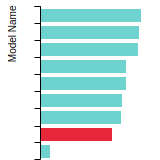
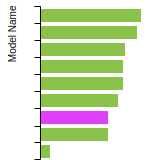
The bar chart shows the number of articles that cited the IP per year. Each bar is divided into green (articles that USED the IP) and blue (articles that did NOT USE the IP).
Users are encouraged to correct Deep Citation errors in determination by clicking the speech icon next to a citing article and providing updated information. This will be integrated into the next Deep Citation learning cycle, which occurs on a regular basis.
OpenKIM acknowledges the support of the Allen Institute for AI through the Semantic Scholar project for providing citation information and full text of articles when available, which are used to train the Deep Citation ML algorithm.
|
This panel provides information on past usage of this interatomic potential (IP) powered by the OpenKIM Deep Citation framework. The word cloud indicates typical applications of the potential. The bar chart shows citations per year of this IP (bars are divided into articles that used the IP (green) and those that did not (blue)). The complete list of articles that cited this IP is provided below along with the Deep Citation determination on usage. See the Deep Citation documentation for more information.
Help us to determine which of the papers that cite this potential actually used it to perform calculations. If you know, click the .
USED (high confidence) H. Amekura et al., “On the mechanism of the shape elongation of embedded nanoparticles,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2020. link Times cited: 9 USED (high confidence) E. Hodille, J. Byggmästar, E. Safi, and K. Nordlund, “Sputtering of beryllium oxide by deuterium at various temperatures simulated with molecular dynamics,” Physica Scripta. 2020. link Times cited: 12 Abstract: The sputtering yield of beryllium oxide (BeO) by incident de… read moreAbstract: The sputtering yield of beryllium oxide (BeO) by incident deuterium (D) ions, for energies from 10 eV to 200 eV, has been calculated for temperatures between 300 K and 800 K using classical molecular dynamics. First, cumulative irradiations are carried out to build up a concentration of D in the material, equal to the experimentally measured concentration, that varies from an atomic fraction of 0.12 (300 K–500 K) to 0.02 (800 K). After building up the concentration of D, non-cumulative irradiations are carried out to estimate the sputtering yields of BeO. For all incident energies, the sputtering yield peaks at 500 K, being closely related to the decrease of the concentration of D above this temperature. At 10 eV, the concentration of D on the surface drives the temperature dependence, while above 30 eV, it is the amount of surface damage created during the cumulative irradiation. read less USED (high confidence) H. Amekura et al., “Vaporlike phase of amorphous
SiO2
is not a prerequisite for the core/shell ion tracks or ion shaping,” Physical Review Materials. 2018. link Times cited: 9 Abstract: The SHI irradiations were performed under the CommonUse Faci… read moreAbstract: The SHI irradiations were performed under the CommonUse Facility Program of JAEA. H.A. was supported by JSPSKAKENHI Grant No. 18K04898. I.S., V.J., F.D., and K.N.
gratefully acknowledge financial support from the Academy
of Finland MESIOS and NANOIS projects, and CPU capacity
grants from the IT Centre for Science CSC in Espoo, Finland. Part of this work was performed at the SAXS/WAXS
beamline at the Australian Synchrotron, part of ANSTO. P.K.
acknowledges the Australian Research Council for financial
support. read less USED (high confidence) I. Gheewala, S. Kenny, and R. Smith, “Atomistic-scale modelling of nanoindentation into optical coatings,” Philosophical Magazine. 2009. link Times cited: 4 Abstract: Simulations of nanoindentation into a typical optical-coatin… read moreAbstract: Simulations of nanoindentation into a typical optical-coatings stack employed in energy efficient glazing have been performed using classical molecular dynamics (MD) and a coupled finite element/MD methodology. The coatings stack consists of a low-emissivity material, Ag, sandwiched between two layers of a transparent conducting oxide (TCO), ZnO. Simulations into both the ZnO and the coatings stack show a strong interaction between the tip symmetry and crystal symmetry in the observed displacement field. A large amount of elastic recovery is observed for both the ZnO system and the coatings stack, but with an impression left on the surface that looks like a crack but extends no further than the tip imprint at maximum depth. The full stack is observed to have a lower hardness once there is a significant penetration of the displacement field into the Ag, when compared to the pure ZnO system. A comparison between the coupled finite element/MD methodology and the fixed boundary MD-only model shows that the boundary conditions have little influence on the calculated results. read less USED (low confidence) B. Agoubi and M. Zemzemi, “Electronic and thermoelectric properties of ZnO/Cu2O heterostructures: First principles calculations,” Ferroelectrics. 2023. link Times cited: 0 Abstract: In this study, we have performed Density Functional Theory c… read moreAbstract: In this study, we have performed Density Functional Theory calculations of ZnO, Cu2O, and ZnO/Cu2O heterostructures. First, we investigated the structural properties of ZnO and the three polymorphs of Cu2O named cubic, tetragonal, and hexagonal. To build a supercell of ZnO/Cu2O, we have justified that the structure of Cu2O(111) which grows on ZnO is a hexagonal structure. Second, we calculated and analyzed the electronic band structures and the density of states of ZnO, Cu2O polymorphs, and ZnO/Cu2O. Finally, to explore the thermoelectric properties of ZnO, Cu2O polymorphs, and ZnO/Cu2O, we combine all Density Functional Theory and Boltzmann transport theory. read less USED (low confidence) S. Burlison, M. Becker, and D. Kovar, “A molecular dynamics study of the effects of velocity and diameter on the impact behavior of zinc oxide nanoparticles,” Modelling and Simulation in Materials Science and Engineering. 2023. link Times cited: 0 Abstract: Molecular dynamics simulations of particle impact have been … read moreAbstract: Molecular dynamics simulations of particle impact have been conducted for a ceramic with mixed ionic-covalent bonding. For these simulations, individual zinc oxide (ZnO) nanoparticles (NPs) were impacted onto a ZnO substrate to observe the effects of impact velocity (1500–3500 m s−1) and particle diameter (10, 20, and 30 nm) on particle deformation and film formation mechanisms that arise during the micro-cold spray process for producing films. The study shows that a critical impact velocity range exists, generally between 1500 and 3000 m s−1, for sticking of the NP to the substrate. Results suggest that solid-state amorphization-induced viscous flow is the primary deformation mechanism present during impact. Decreasing particle diameter and increasing impact velocity results in an increased degree of amorphization and higher local temperatures within the particle. The impact behavior of mixed ionic-covalent bonded ZnO is compared to the behavior of previously studied ionic and covalent materials. read less USED (low confidence) B. Yao, Z. R. Liu, D. Legut, and R. F. Zhang, “Hybrid potential model with high feasibility and flexibility for metallic and covalent solids,” Physical Review B. 2023. link Times cited: 0 USED (low confidence) M. Moin et al., “A comprehensive correlated analysis of Ra-Doped (ZnO_2, ZnO) for optoelectronic applications: a first-principle study,” Journal of Molecular Modeling. 2023. link Times cited: 0 USED (low confidence) G. Clavier and A. Thompson, “Computation of the thermal elastic constants for arbitrary manybody potentials in LAMMPS using the stress-fluctuation formalism,” Comput. Phys. Commun. 2023. link Times cited: 1 USED (low confidence) K. Ren, L. Liu, J. Li, H. Pan, and Z. Wang, “Lubrication Behavior of n-hexadecane on ZnO Layer at the Nanoscale: A Molecular Dynamic Exploration,” Tribology Letters. 2022. link Times cited: 1 USED (low confidence) E. Hodille, J. Byggmästar, Y. Ferro, and K. Nordlund, “Molecular dynamics study of hydrogen isotopes at the Be/BeO interface,” Journal of Physics: Condensed Matter. 2022. link Times cited: 3 Abstract: Molecular dynamics simulations are used to investigate the b… read moreAbstract: Molecular dynamics simulations are used to investigate the behaviour of D atoms at two interfaces between beryllium (Be) and beryllium oxide (BeO). After relaxation of the simulation cell, there are (a) localised defects at the interface and (b) a hexagonal misfit dislocation network creating a succession of compressed and expanded area from each side of the interface. The simulations between 750 K and 1500 K for tens to hundreds of nanoseconds show that both interfaces act as trapping sites for D atoms. The simulations also show that D atoms tend to migrate in the material where the hydrogen isotope solubility is the highest as predicted by thermodynamics. However, the simulations also shows that there are additional kinetic barriers (D trapping sites, D2 formation/dissociation in BeO) that slow down the path to equilibrium. These additional kinetic barriers may influence the fuel retention and permeation in Be materials. read less USED (low confidence) X. Zhu and X. Cheng, “Molecular dynamics study of tilt grain boundary evolution during the growth of beryllium thin films,” Journal of Crystal Growth. 2020. link Times cited: 0 USED (low confidence) G. Plummer and G. Tucker, “Bond-order potentials for theTi3AlC2andTi3SiC2MAX phases,” Physical Review B. 2019. link Times cited: 12 USED (low confidence) J. Byggmästar, M. J. Nagel, K. Albe, K. Henriksson, and K. Nordlund, “Analytical interatomic bond-order potential for simulations of oxygen defects in iron,” Journal of Physics: Condensed Matter. 2019. link Times cited: 11 Abstract: We present an analytical bond-order potential for the Fe–O s… read moreAbstract: We present an analytical bond-order potential for the Fe–O system, capable of reproducing the basic properties of wüstite as well as the energetics of oxygen impurities in -iron. The potential predicts binding energies of various small oxygen-vacancy clusters in -iron in good agreement with density functional theory results, and is therefore suitable for simulations of oxygen-based defects in iron. We apply the potential in simulations of the stability and structure of Fe/FeO interfaces and FeO precipitates in iron, and observe that the shape of FeO precipitates can change due to formation of well-defined Fe/FeO interfaces. The interface with crystalline Fe also ensures that the precipitates never become fully amorphous, no matter how small they are. read less USED (low confidence) E. Hodille, J. Byggmästar, E. Safi, and K. Nordlund, “Molecular dynamics simulation of beryllium oxide irradiated by deuterium ions: sputtering and reflection,” Journal of Physics: Condensed Matter. 2019. link Times cited: 10 Abstract: The sputtering and reflection properties of wurtzite berylli… read moreAbstract: The sputtering and reflection properties of wurtzite beryllium oxide (BeO) subjected to deuterium (D) ions bombardment at 300 K with ion energy between 10 eV and 200 eV is studied by classical molecular dynamics. Cumulative irradiations of wurtzite BeO show a D concentration threshold above which an ‘unphysical dramatic’ sputtering is observed. From the cumulative irradiations, simulation cells with different D concentrations are used to run non-cumulative irradiations at different concentrations. Using a D concentration close to the experimentally determined saturation concentration (0.12 atomic fraction), the simulations are able to reproduce accurately the experimental sputtering yield of BeO materials. The processes driving the sputtering of beryllium (Be) and oxygen (O) atoms as molecules are subsequently determined. At low irradiation energy, between 10 eV and 80 eV, swift chemical sputtering (SCS) is dominant and produces mostly ODz molecules. At high energy, the sputtered molecules are mostly BexOy molecules (mainly BeO dimer). Four different processes are associated to the formation of such molecules: the physical sputtering of BeO dimer, the delayed SCS not involving D ions and the detachment-induced sputtering. The physical sputtering of BeO dimer can be delayed if the sputtering event implies two interactions with the incoming ion (first interaction in its way in the material, the other in its way out if it is backscattered). The detachment-induced sputtering is a characteristic feature of the ‘dramatic’ sputtering and is mainly observed when the concentration of D is close to the threshold leading to this sputtering regime. read less USED (low confidence) M. Mock and K. Albe, “Modelling of dislocation-solute interaction in ODS steels: Analytic bond-order potential for the iron-yttrium system,” Journal of Nuclear Materials. 2018. link Times cited: 6 USED (low confidence) L. S. I. Liyanage et al., “From Electrons to Atoms: Designing an Interatomic Potential for Fe-C Alloys.” 2018. link Times cited: 0 USED (low confidence) Y. Chergui, T. Aouaroun, M. Hadley, R. Belkada, R. Chemam, and D. Mekki, “Molecular dynamics simulation of ZnO wurtzite phase under high and low pressures and temperatures,” Materials Research Express. 2017. link Times cited: 1 Abstract: Isothermal and isobaric ensembles behaviours of ZnO wurtzite… read moreAbstract: Isothermal and isobaric ensembles behaviours of ZnO wurtzite phase have been investigated, by parallel molecular dynamics method and using Buckingham potential, which contains long-range Coulomb, repulsive exponential, and attractive dispersion terms. To conduct our calculations, we have used dl_poly 4 software, under which the method is implemented. We have examined the influence of the temperature and pressure on molar volume in the ranges of 300–3000 K and 0–200 GPa. Isothermal-isobaric relationships, fluctuations, standard error, equilibrium time, molar volume and its variation versus time are predicted and analyzed. Our results are close to available experimental data and theoretical results. read less USED (low confidence) K. Albe, P. Ágoston, and J. Pohl, “Ab InitioModeling of Defects in Semiconductors.” 2016. link Times cited: 0 USED (low confidence) Y. Liu, M. Shahzad, and Y. Qi, “Growth of a-axis ZnO films on the defective substrate with different O/Zn ratios: A reactive force field based molecular dynamics study,” Journal of Alloys and Compounds. 2015. link Times cited: 7 USED (low confidence) T. Liang et al., “Classical atomistic simulations of surfaces and heterogeneous interfaces with the charge-optimized many body (COMB) potentials,” Materials Science & Engineering R-reports. 2013. link Times cited: 207 USED (low confidence) Y.-T. Cheng et al., “Atomistic simulations of the adsorption and migration barriers of Cu adatoms on ZnO surfaces using COMB potentials,” Surface Science. 2012. link Times cited: 25 USED (low confidence) D. Zagorac, J. C. Schön, and M. Jansen, “Energy Landscape Investigations Using the Prescribed Path Method in the ZnO System,” Journal of Physical Chemistry C. 2012. link Times cited: 29 Abstract: An important issue in modern solid-state chemistry and nanot… read moreAbstract: An important issue in modern solid-state chemistry and nanotechnology is the development of a general methodology to predict possible (meta)stable crystalline and nanocrystalline modifications and to study the possible transition routes among them. To analyze the stability of the various potential modifications and study the possible low energy paths among them, the so-called prescribed path method is employed. This method allows us to explore transition routes and barriers between even distant minima, suggesting possible transition states and specific transition paths for more detailed analysis, as well as to gain more insights into the temperature dependence of the synthesis and transformation processes in the system. In this study, we describe and employ the prescribed path method for the example of the energy landscape of ZnO. The focus is on the influence of the temperature on the transformations along the path and on the stability of the various structures in the ZnO system, such as the wurtzite-, t... read less USED (low confidence) P. M. Diehm, P. Ágoston, and K. Albe, “Size-dependent lattice expansion in nanoparticles: reality or anomaly?,” Chemphyschem : a European journal of chemical physics and physical chemistry. 2012. link Times cited: 180 Abstract: Size-dependent lattice expansion of nanoparticles is observe… read moreAbstract: Size-dependent lattice expansion of nanoparticles is observed for many ionic compounds, including metal oxides, while lattice contraction prevails for pure metals. However, the physical origin of this effect, which is of importance for the thermodynamic, chemical and electronic properties of nanoparticles, is discussed controversially. After a survey of the experimental literature, revealing a wide variety of materials with size-dependent lattice expansion, we show that the negative surface stress is the key reason for lattice expansion, while the excess of lattice sums or point defects of various charge states can be excluded as general explanations. Ab initio calculations of surface stresses for various surface structures of metal oxides confirm the model of a surface-induced lattice expansion. read less USED (low confidence) K. Albe, P. Ágoston, and J. Pohl, “Ab‐Initio Modeling of Defects in Semiconductors.” 2011. link Times cited: 1 USED (low confidence) O. Pakarinen, F. Djurabekova, and K. Nordlund, “Density evolution in formation of swift heavy ion tracks in insulators,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2010. link Times cited: 27 USED (low confidence) S. Deulkar, H. Yo, and J.-L. Huang, “Tem-based investigations on CVD-assisted growth of ZnO nanowires inside nanochannels of anodized aluminum Oxide template,” International Journal of Nanoscience. 2010. link Times cited: 1 Abstract: The role of Chemical Vapor Deposition's (CVD) temperatu… read moreAbstract: The role of Chemical Vapor Deposition's (CVD) temperature and time duration on the mass transport properties and repercussions of these properties on the crystalline growth of ZnO nanowires inside Anodic Aluminum Oxide (AAO) nanotubes (aspect ratio = 1500) have been investigated. CVD assisted growth of nanowires in AAO nanochannels was carried out for 27 min at deposition temperature of 813 K (A1) and for 180 min at 903 K (A2) under identical gas flow rates (Ar: 23 sccm and O2: 0.45 sccm) under a chamber pressure of 4.45 torr. Electron diffraction patterns (SAED) of the ZnO diffused inside A1 nanotube (exposed in Focused Ion Beam milled cross-sections) showed a ringed pattern, signifying polycrystalline morphology at the tip as well as the channel bottom. For A2, a single crystal formation at least in the tip region was confirmed by the SAED pattern while the corresponding High Resolution TEM image (HRTEM) revealed a lattice spacing of 0.24 nm corresponding to the (1 0 1) growth direction of zincite (JCPDS-36-1451). Chemical composition of the nanowires, reflected the inherent stoichiometry of preferentially nucleated ZnO in the grooved orifices of individual nanopores. Data points of normalised values of the Zn concentration (TEM-EDS line scan) versus penetration depth (~ 0.6 μm), were empirically fitted with solutions of modified Fickian diffusion equation in 1-D. The Diffusion coefficient values of the order of 0.06 nm2 s-1 and 0.22 nm2 s-1 were obtained for ZnO flow inside nanochannels of A1 and A2, respectively. Observed variations, in diffusivity and relative density, have been explained qualitatively by considering the transport of ZnO inside the nanochannels as arising from superposition of the temperature dependent contributions of the convectional viscous creep and surface diffusion components. read less USED (low confidence) T. E. Mølholt et al., “Temperature and dose dependence of defect complex formation with ion implanted Mn/Fe in ZnO,” Physica B-condensed Matter. 2009. link Times cited: 15 USED (low confidence) B. Coasne, A. Mezy, R. Pellenq, D. Ravot, and J. Tedenac, “Zinc oxide nanostructures confined in porous silicas.,” Journal of the American Chemical Society. 2009. link Times cited: 18 Abstract: We report on molecular simulations of zinc oxide nanostructu… read moreAbstract: We report on molecular simulations of zinc oxide nanostructures obtained within silica nanopores of diameter D = 1.6 nm and D = 3.2 nm. Both the effects of confinement (by varying the pore size) and degree of pore filling on the structure of the nanomaterial are addressed. Two complementary approaches are adopted: 1) the stability of the three crystalline phases of ZnO (wurtzite, rocksalt, and blende) in the silica nanopores is studied, and 2) ZnO nanostructures are obtained by slowly cooling down a homogeneous liquid phase confined in the silica pores. None of the ideal nanostructures (wurtzite, rocksalt, blende) retains the ideal structure of the initial crystal when confined within the silica pores. Only the structure starting from the ideal wurtzite nanocrystal remains significantly crystalline after relaxation, as revealed by the marked peaks in the pair correlation functions for this system. The morphology and degree of cristallinity of the structures are found to depend on the parameters involved in the synthesis (pore size, filling density). Nanograin boundaries are observed between domains of different crystal structures. Reminiscent features of the bulk behavior, such as faceting of the nanostructures, are also observed when the system size becomes large. We show that the use of nanopores as a template imposes that the confined particles exhibit neutral (basal) surfaces. These predictions provide a guide to experiments on semiconductor nanoparticles. read less USED (low confidence) C. R. Catlow, S. French, A. Sokol, A. Al-Sunaidi, and S. Woodley, “Zinc oxide: A case study in contemporary computational solid state chemistry,” Journal of Computational Chemistry. 2008. link Times cited: 101 Abstract: Computational techniques have been applied to study a broad … read moreAbstract: Computational techniques have been applied to study a broad range of chemical and physical properties of zinc oxide. Both interatomic‐potential and density functional theory methods are used to investigate structural, thermodynamic, surface, and defect properties. We survey the structures and energies of nano‐particulate zinc oxide. © 2008 Wiley Periodicals, Inc. J Comput Chem 2008 read less USED (low confidence) J. Wróbel and J. Piechota, “On the structural stability of ZnO phases,” Solid State Communications. 2008. link Times cited: 38 USED (low confidence) K. Nordlund and S. Dudarev, “Interatomic potentials for simulating radiation damage effects in metals,” Comptes Rendus Physique. 2008. link Times cited: 29 USED (low confidence) M. Müller, P. Erhart, and K. Albe, “Thermodynamics of L 1 0 ordering in FePt nanoparticles studied by Monte Carlo simulations based on an analytic bond-order potential,” Physical Review B. 2007. link Times cited: 64 Abstract: The size dependence of the order-disorder transition in FePt… read moreAbstract: The size dependence of the order-disorder transition in FePt nanoparticles with an $L{1}_{0}$ structure is investigated by means of Monte Carlo simulations based on an analytic bond-order potential for FePt. A cross parametrization for the Fe-Pt interaction is proposed, which complements existing potentials for the constituents Fe and Pt. This FePt potential properly describes structural properties of ordered and disordered phases, surface energies, and the $L{1}_{0}$ to $A1$ transition temperature in bulk FePt. The potential is applied for examining the ordering behavior in small particles. The observed lowering of the order-disorder transition temperature with decreasing particle size confirms previous lattice-based Monte Carlo simulations [M. M\"uller and K. Albe, Phys. Rev. B 72, 094203 (2005)]. Although a distinctly higher amount of surface induced disorder is found in comparison to previous studies based on lattice-type Hamiltonians, the presence of lattice strain caused by the tetragonal distortion of the $L{1}_{0}$ structure does not have a significant influence on the depression of the ordering temperature with decreasing particle size. read less USED (low confidence) S. M. Zamzamian, S. Feghhi, and M. Samadfam, “Theoretical and computational investigation on the radiation-induced point defects in cementite: Picosecond timescale,” Journal of Nuclear Materials. 2021. link Times cited: 1 USED (low confidence) R. Evarestov, “Binary Oxides of Transition Metals: ZnO, TiO$_2, ZrO_2, HfO_2$.” 2020. link Times cited: 0 USED (low confidence) K. Ellmer, “Transparent Conductive Zinc Oxide and Its Derivatives.” 2011. link Times cited: 46 NOT USED (low confidence) D. Raymand, A. Duin, M. Baudin, and K. Hermansson, “A reactive force field (ReaxFF) for zinc oxide,” Surface Science. 2008. link Times cited: 127 NOT USED (low confidence) R. Jones, C. Weinberger, S. Coleman, and G. Tucker, “Introduction to Atomistic Simulation Methods.” 2016. link Times cited: 1 NOT USED (high confidence) J. Goniakowski, S. Menon, G. Laurens, and J. Lam, “Nonclassical Nucleation of Zinc Oxide from a Physically Motivated Machine-Learning Approach,” The Journal of Physical Chemistry C. 2021. link Times cited: 3 Abstract: Observing non-classical nucleation pathways remains challeng… read moreAbstract: Observing non-classical nucleation pathways remains challenging in simulations of complex materials with technological interests. This is because it requires very accurate force fields that can capture the whole complexity of their underlying interatomic interactions and an advanced structural analysis. Here, we first report the construction of a machine-learning force field for zinc oxide interactions using the Physical LassoLars Interaction Potentials approach which allows us to be predictive even for untrained structures. Then, we carried out freezing simulations from a liquid and observed the crystal formation with atomistic precision. Our results, which are analyzed using a data-driven approach based on bond order parameters, demonstrate the presence of both prenucleation clusters and two-step nucleation scenarios thus retrieving seminal predictions of non-classical nucleation pathways made on much simpler models. read less NOT USED (high confidence) Z. Fan, Y. Wang, X. Gu, P. Qian, Y. Su, and T. Ala‐Nissila, “A minimal Tersoff potential for diamond silicon with improved descriptions of elastic and phonon transport properties,” Journal of Physics: Condensed Matter. 2019. link Times cited: 10 Abstract: Silicon is an important material and many empirical interato… read moreAbstract: Silicon is an important material and many empirical interatomic potentials have been developed for atomistic simulations of it. Among them, the Tersoff potential and its variants are the most popular ones. However, all the existing Tersoff-like potentials fail to reproduce the experimentally measured thermal conductivity of diamond silicon. Here we propose a modified Tersoff potential and develop an efficient open source code called GPUGA (graphics processing units genetic algorithm) based on the genetic algorithm and use it to fit the potential parameters against energy, virial and force data from quantum density functional theory calculations. This potential, which is implemented in the efficient open source GPUMD (graphics processing units molecular dynamics) code, gives significantly improved descriptions of the thermal conductivity and phonon dispersion of diamond silicon as compared to previous Tersoff potentials and at the same time well reproduces the elastic constants. Furthermore, we find that quantum effects on the thermal conductivity of diamond silicon at room temperature are non-negligible but small: using classical statistics underestimates the thermal conductivity by about 10% as compared to using quantum statistics. read less NOT USED (high confidence) X. Zhu and Y. Lu, “Growth of beryllium thin films on beryllium (0001) surface: Influence of incident energy and incident angle by molecular dynamics simulation,” Journal of Applied Physics. 2018. link Times cited: 5 Abstract: The morphology and microstructure of metallic thin films syn… read moreAbstract: The morphology and microstructure of metallic thin films synthesized by magnetron sputtering deposition are sensitive to incident energy and incident angle. The role of incident energy and incident angle in films’ morphology evolution of the beryllium thin films’ growth on beryllium (0001) surface was studied by molecular dynamics simulations. The analytical bond order potential was used to represent the interatomic interactions, and the common neighbor analysis algorithm for crystal structures was used for the structural characterization of the simulated films. It is found that when the incident energy is between 1 eV and 20 eV, the increased incident energy is beneficial to grow uniform crystal films and, when the incident energy is greater than 15 eV, the interstitial atoms formed inside the films. Furthermore, under the small incident angle conditions, the morphology of a smooth surface was formed, which means that the vertical incident conditions are desired for the growth of high quality films. In short, vertically inserted atoms with hyperthermal energy (5–10 eV) are more propitious for the growth of perfect crystal Be thin films. The obtained results can be used to guide the experiment. read less NOT USED (high confidence) J. Byggmästar, E. Hodille, Y. Ferro, and K. Nordlund, “Analytical bond order potential for simulations of BeO 1D and 2D nanostructures and plasma-surface interactions,” Journal of Physics: Condensed Matter. 2018. link Times cited: 18 Abstract: An analytical interatomic bond order potential for the Be–O … read moreAbstract: An analytical interatomic bond order potential for the Be–O system is presented. The potential is fitted and compared to a large database of bulk BeO and point defect properties obtained using density functional theory. Its main applications include simulations of plasma-surface interactions involving oxygen or oxide layers on beryllium, as well as simulations of BeO nanotubes and nanosheets. We apply the potential in a study of oxygen irradiation of Be surfaces, and observe the early stages of an oxide layer forming on the Be surface. Predicted thermal and elastic properties of BeO nanotubes and nanosheets are simulated and compared with published ab initio data. read less NOT USED (high confidence) A. Galashev and O. Rakhmanova, “Computer study the oxygen release from Al melts,” Modelling and Simulation in Materials Science and Engineering. 2018. link Times cited: 1 Abstract: The behavior of oxygen ions in the Al melts under action of … read moreAbstract: The behavior of oxygen ions in the Al melts under action of a constant electric field was studied by molecular dynamics. The rate of O2− ions moving up from the cathode to the melt surface increases. The time of the first ion reaching the surface decreases with increase in O2− concentration. The Al and O2− self-diffusion coefficients increase with increasing concentration of ions in the system. The structure of the neighborhood of oxygen ions was studied in detail by statistical geometry. The distributions of truncated Voronoi polyhedra according to the number of faces and of faces according to the number of sides were determined. Simplified polyhedra were obtained after elimination of small-scale thermal fluctuations from the model. The picture of the oxygen ions final location can vary greatly depending on the boundary conditions and their application sequence. read less NOT USED (high confidence) B. Narayanan et al., “Machine learnt bond order potential to model metal-organic (Co-C) heterostructures.,” Nanoscale. 2017. link Times cited: 7 Abstract: A fundamental understanding of the inter-relationships betwe… read moreAbstract: A fundamental understanding of the inter-relationships between structure, morphology, atomic scale dynamics, chemistry, and physical properties of mixed metallic-covalent systems is essential to design novel functional materials for applications in flexible nano-electronics, energy storage and catalysis. To achieve such knowledge, it is imperative to develop robust and computationally efficient atomistic models that describe atomic interactions accurately within a single framework. Here, we present a unified Tersoff-Brenner type bond order potential (BOP) for a Co-C system, trained against lattice parameters, cohesive energies, equation of state, and elastic constants of different crystalline phases of cobalt as well as orthorhombic Co2C derived from density functional theory (DFT) calculations. The independent BOP parameters are determined using a combination of supervised machine learning (genetic algorithms) and local minimization via the simplex method. Our newly developed BOP accurately describes the structural, thermodynamic, mechanical, and surface properties of both the elemental components as well as the carbide phases, in excellent accordance with DFT calculations and experiments. Using our machine-learnt BOP potential, we performed large-scale molecular dynamics simulations to investigate the effect of metal/carbon concentration on the structure and mechanical properties of porous architectures obtained via self-assembly of cobalt nanoparticles and fullerene molecules. Such porous structures have implications in flexible electronics, where materials with high electrical conductivity and low elastic stiffness are desired. Using unsupervised machine learning (clustering), we identify the pore structure, pore-distribution, and metallic conduction pathways in self-assembled structures at different C/Co ratios. We find that as the C/Co ratio increases, the connectivity between the Co nanoparticles becomes limited, likely resulting in low electrical conductivity; on the other hand, such C-rich hybrid structures are highly flexible (i.e., low stiffness). The BOP model developed in this work is a valuable tool to investigate atomic scale processes, structure-property relationships, and temperature/pressure response of Co-C systems, as well as design organic-inorganic hybrid structures with a desired set of properties. read less NOT USED (high confidence) A. Bandura, R. Evarestov, S. I. Lukyanov, S. Piskunov, and Y. Zhukovskii, “Simulation of Young’s moduli for hexagonal ZnO [0 0 0 1]-oriented nanowires: first principles and molecular mechanical calculations,” Materials Research Express. 2017. link Times cited: 10 Abstract: Morphologically reproducible wurtzite-structured zinc oxide … read moreAbstract: Morphologically reproducible wurtzite-structured zinc oxide nanowires (ZnO NWs) can be synthesized by different methods. Since ZnO NWs have been found to possess piezoelectricity, a comprehensive study of their mechanical properties, e.g. deformations caused by external compression or stretching, is one of the actual tasks of this paper. We have calculated wurtzite-structured [0 0 0 1]-oriented ZnO NWs whose diameters have been varied within 1–5 nm and 1–20 nm ranges when using either ab initio (hybrid DFT-LCAO) or force-field (molecular mechanical) methods, respectively (the minimum diameter dNW of experimentally synthesized NWs has been estimated on average to be ~20 nm). When using both chosen calculation approaches, the values of Young’s moduli determined for the mentioned ranges of NW diameters have been found to be qualitatively compatible (168–169 GPa for 5 nm NW thickness), whereas results of molecular mechanical simulations on YNW for 20 nm-thick NWs (160–162 GPa) have been qualitatively comparable with those experimentally measured along the [0 0 0 1] direction of NW loading. In all the cases, a gradual increase of the NW diameter has resulted in an asymptotic decrease of Young’s modulus consequently approaching that (Yb) of wurtzite-structured ZnO bulk along its [0 0 0 1] axis. The novelty of this study is that we combine the computation methods of quantum chemistry and molecular mechanics, while the majority of previous studies with the same aim have focused on the application of different classical molecular dynamical methods. read less NOT USED (high confidence) O. Turkay, H. Inan, and A. Dimoglo, “Experimental and theoretical study on catalytic ozonation of humic acid by ZnO catalyst,” Separation Science and Technology. 2017. link Times cited: 9 Abstract: ABSTRACT Ozonation and catalytic ozonation of humic acid (HA… read moreAbstract: ABSTRACT Ozonation and catalytic ozonation of humic acid (HA) in the presence of ZnO were examined in a batch scale reactor. The degradation of HA by catalytic ozonation was found to be much more effective than ozonation alone. The quantum chemistry calculations showed that the reaction of the O3 disproportionation on the surface of ZnO corresponds to the barrierless mechanism. The activation energy of the transition state formation was −5.25 eV. The active oxygen atom formed on the surface of ZnO was found as interacting with both water molecules and dissolved organic molecules, which might lead to further oxidizing reactions. read less NOT USED (high confidence) A. Stukowski, E. Fransson, M. Mock, and P. Erhart, “Atomicrex—a general purpose tool for the construction of atomic interaction models,” Modelling and Simulation in Materials Science and Engineering. 2017. link Times cited: 17 Abstract: We introduce atomicrex, an open-source code for constructing… read moreAbstract: We introduce atomicrex, an open-source code for constructing interatomic potentials as well as more general types of atomic-scale models. Such effective models are required to simulate extended materials structures comprising many thousands of atoms or more, because electronic structure methods become computationally too expensive at this scale. atomicrex covers a wide range of interatomic potential types and fulfills many needs in atomistic model development. As inputs, it supports experimental property values as well as ab initio energies and forces, to which models can be fitted using various optimization algorithms. The open architecture of atomicrex allows it to be used in custom model development scenarios beyond classical interatomic potentials while thanks to its Python interface it can be readily integrated e.g., with electronic structure calculations or machine learning algorithms. read less NOT USED (high confidence) Z. Fan, “Molecular Simulations of Nanoscale Transformations in Ionic Semiconductor Nanocrystals.” 2016. link Times cited: 0 Abstract: The aim of the study described in this thesis is to obtain a… read moreAbstract: The aim of the study described in this thesis is to obtain a profound understanding of transformations in NCs at the atomic level, by performing molecular simulations for such transformations, and by comparing the simulation results with available experimental high resolution transmission electron microscopy (HRTEM) data to validate the simulations and to reveal underlying physical mechanisms. These transformations include structural and morphological transitions and cation exchange processes in ionic nanocrystals (II-VI and IV-VI semiconductors). The main simulation method used is classical Molecular Dynamics (MD) simulation. First principles density functional theory (DFT) calculations were used to develop empirical force fields that are able to accurately reproduce phase transitions. Using these newly developed force fields, large scaled classical MD simulations were carried out and linked to HRTEM experiments. The partially charged rigid ion model (PCRIM) was chosen for the force fields. This PCRIM approach has a simple functional form with a few number of parameters and has a clear physical meaning for ionic crystals. To simulate cation exchange in colloidal NC systems at the NC/solution interface, we used a combination of all-atom force fields and a coarse-grained model. In Chapter 2, an ab-initio based force field for ZnO is developed within the framework of the PCRIM approach. The values of the partial charges were determined by Bader charge analysis of DFT calculations on various ZnO phases. Beside Coulombic interactions, only short-ranged pairwise interatomic interactions were included. An initial guess of the parameters of the short-ranged pair potentials were first obtained by the lattice inversion method. The parameters were further adjusted by an ab-initio potential surface fitting procedure. The new ZnO force field has a very simple functional form is able to accurately reproduce several important physical properties of ZnO materials. These physical properties include the lattice parameters and phase stability of several ZnO polymorphs, as well as the elastic constants, bulk moduli, phonon dispersion, and melting points of wurtzite ZnO. The transition pressure of the wurtzite-to-rocksalt transition calculated with the force field equals 12.3 GPa, in agreement with experimental measurements and DFT calculations. A wurtzite-to-honeycomb phase transition is predicted at an uniaxial pressure of 8.8 GPa. We found a rational and effective way to derive force fields with simple functional forms for accurate simulations of phase transitions in ionic crystals. In Chapter 3, we developed a transferable force field for CdS-CdSe-PbSPbSe solid systems. The selection of the force field and the fitting procedure are similar to that of the ZnO force field in Chapter 2. The challenges when developing this force field were to maintain the transferability of this force field for four materials (CdS, CdSe, PbS, and PbSe) and to describe their mixed phases. This was solved by assuming that different cations/anions have the same values of the partial charges, and that shortranged interatomic interactions between two cations/anions are the same in different materials. For the mixed phases, DFT calculations of the mixed phases were included in both the training and validation sets. This work is the first step for further simulation studies of these II-VI and IV-VI semiconductor NCs and heteronanocrystals (HNCs). In Chapter 4, a thermally induced morphological and structural transition of CdSe NCs was investigated using MD simulations. In MD simulations, a CdSe nanosphere with the ZB structure transforms into a tetrapodlike morphology at 800 K. In a CdSe tetrapod, four WZ legs attach to the {111} facets of a tetrahedral ZB core. This transformation is achieved by a layer-by-layer slip of the ZB-{111} bilayer. Simulations show that the slips are mediated by the formation of Cd vacancies on the surface of the NCs to overcome the potentially large energy barriers associated with slip. The morphology of the annealed NCs is found to be temperature and size-dependent. An octapod-like morphology is found in NCs with a relatively large NC size and in a certain range of the heating temperature. Surprisingly, nanoscale transformations of CdSe NCs have been directly observed in HRTEM in situ heating experiments. Our findings provide a simple method to modify the morphology of ionic NCs and can potentially be used in the synthesis of branched NCs. The cation exchange process of PbSe/CdSe HNCs has been investigated by HRTEM in situ heating experiments in combination with MD simulations and DFT calculations in Chapter 5. In the HRTEM experiments, we bserved that Cd atoms in PbSe/CdSe nanodumbbells (CdSe rods with one or two PbSe tip(s)) are replaced by Pb atoms. The exchange rate depends on the heating temperature and the amount of Pb atoms present in the system. Sometimes, fully converted PbSe nanodumbells can be observed. MD simulations were performed to investigate the mechanism of this cation exchange process. It was found that the the CdSe domains near the PbSe/CdSe interfaces have significant structural disorder. These findings are in line with the experimental observation that the exchange process proceeds in a layer-by-layer fashion along the WZ- direction. We concluded that cation exchange in PbSe/CdSe HNCs is mediated by the local structural disorder which enables the formation of vacancies and accelerated the motion of cations. In Chapter 6, a coarse-grained psuedoligand model was introduced to simulate cation exchange in PbS colloidal NCs taking into account the cation-solvent interactions. Modelling colloidal NC systems including interactions with the solvent has long been a challenge due to the large system size and long time scales. Here, we incorporated the effects of ligands and solvents into negatively charged large spherical coarse-grained psuedoligands. MD simulations combining coarse-grained and all-atom models can successfully reproduce the cation exchange process in PbS colloidal NCs. Simulations show that the exchange rate and system equilibrium can be controlled by the temperature and by changing ligands. The exchange process is directly related to vacancy formation and the high mobility of Cd ions at the PbS/CdS interface. Our simulations also predict that high-pressure conditions will be beneficial for achieving fast exchange at elevated temperatures. Our coarse-grained model can be easily extended to other systems for the computational investigation of transformations in nanostructures. read less NOT USED (high confidence) B. Narayanan et al., “Describing the Diverse Geometries of Gold from Nanoclusters to Bulk—A First-Principles-Based Hybrid Bond-Order Potential,” Journal of Physical Chemistry C. 2015. link Times cited: 27 Abstract: Molecular dynamics simulations using empirical force fields … read moreAbstract: Molecular dynamics simulations using empirical force fields (EFFs) are crucial for gaining fundamental insights into atomic structure and long time scale dynamics of Au nanoclusters with far-reaching applications in energy and devices. This approach is thwarted by the failure of currently available EFFs in describing the size-dependent dimensionality and diverse geometries exhibited by Au clusters (e.g., planar structures, hollow cages, tubes, pyramids, space-filled structures). Here, we mitigate this issue by introducing a new hybrid bond-order potential (HyBOP), which accounts for (a) short-range interactions via Tersoff-type BOP terms that accurately treat bond directionality and (b) long-range dispersion effects by a scaled Lennard–Jones term whose contribution depends on the local atomic density. We optimized the independent parameters for our HyBOP using a global optimization scheme driven by genetic algorithms. Moreover, to ensure good transferability of these parameters across different length sca... read less NOT USED (high confidence) T. Burian et al., “Soft x-ray free-electron laser induced damage to inorganic scintillators,” Optical Materials Express. 2015. link Times cited: 12 Abstract: An irreversible response of inorganic scintillators to inten… read moreAbstract: An irreversible response of inorganic scintillators to intense soft x-ray laser radiation was investigated at the FLASH (Free-electron LASer in Hamburg) facility. Three ionic crystals, namely, Ce:YAG (cerium-doped yttrium aluminum garnet), PbWO4 (lead tungstate), and ZnO (zinc oxide), were exposed to single 4.6 nm ultra-short laser pulses of variable pulse energy (up to 12 μJ) under normal incidence conditions with tight focus. Damaged areas produced with various levels of pulse fluences, were analyzed on the surface of irradiated samples using differential interference contrast (DIC) and atomic force microscopy (AFM). The effective beam area of 22.2 ± 2.2 μm2 was determined by means of the ablation imprints method with the use of poly(methyl methacrylate) - PMMA. Applied to the three inorganic materials, this procedure gave almost the same values of an effective area. The single-shot damage threshold fluence was determined for each of these inorganic materials. The Ce:YAG sample seems to be the most radiation resistant under the given irradiation conditions, its damage threshold was determined to be as high as 660.8 ± 71.2 mJ/cm2. Contrary to that, the PbWO4 sample exhibited the lowest radiation resistance with a threshold fluence of 62.6 ± 11.9 mJ/cm2. The threshold for ZnO was found to be 167.8 ± 30.8 mJ/cm2. Both interaction and material characteristics responsible for the damage threshold difference are discussed in the article. read less NOT USED (high confidence) K. Nordlund, C. Björkas, T. Ahlgren, A. Lasa, and A. Sand, “Multiscale modelling of plasma–wall interactions in fusion reactor conditions,” Journal of Physics D: Applied Physics. 2014. link Times cited: 58 Abstract: The interaction of fusion reactor plasma with the material o… read moreAbstract: The interaction of fusion reactor plasma with the material of the first wall involves a complex multitude of interlinked physical and chemical effects. Hence, modern theoretical treatment of it relies to a large extent on multiscale modelling, i.e. using different kinds of simulation approaches suitable for different length and time scales in connection with each other. In this review article, we overview briefly the physics and chemistry of plasma–wall interactions in tokamak-like fusion reactors, and present some of the most commonly used material simulation approaches relevant for the topic. We also give summaries of recent multiscale modelling studies of the effects of fusion plasma on the modification of the materials of the first wall, especially on swift chemical sputtering, mixed material formation and hydrogen isotope retention in tungsten. read less NOT USED (high confidence) S. Wang et al., “New Ab Initio Based Pair Potential for Accurate Simulation of Phase Transitions in ZnO,” Journal of Physical Chemistry C. 2014. link Times cited: 42 Abstract: A set of interatomic pair potentials is developed for ZnO ba… read moreAbstract: A set of interatomic pair potentials is developed for ZnO based on the partially charged rigid ion model (PCRIM). The derivation of the potentials combines lattice inversion, empirical fitting, and ab initio energy surface fitting. We show that, despite the low number of parameters in this model (8), a wide range of physical properties is accurately reproduced using the new potential model. The calculated lattice parameters and elastic constants of ZnO in the wurtzite (WZ) phase, as well as the lattice parameters and stabilities of ZnO in other high-pressure and metastable phases, agree well with experiments and with density functional theory (DFT) calculations. The calculated transition pressure of the wurtzite-to-rocksalt (WZ-to-RS) transition is 12.3 GPa. A wurtzite-to-honeycomb (WZ-to-HC) phase transition induced by uniaxial pressure along the c-axis is simulated by means of molecular dynamics (MD) simulations. The WZ-to-HC transition takes place at an uniaxial pressure of 8.8 GPa while the reverse tr... read less NOT USED (high confidence) K. Nordlund and F. Djurabekova, “Multiscale modelling of irradiation in nanostructures,” Journal of Computational Electronics. 2014. link Times cited: 42 NOT USED (high confidence) K. Li, W. Yang, J. Wei, S. Du, and Y. Li, “Modeling of metal–oxide semiconductor: Analytical bond-order potential for cupric oxide,” Chinese Physics B. 2014. link Times cited: 0 Abstract: Atomistic potentials for cupric element and cupric oxide are… read moreAbstract: Atomistic potentials for cupric element and cupric oxide are derived based on the analytical bond-order scheme that was presented by Brenner [Brenner D W, "Erratum: Empirical potential for hydrocarbons for use in simulating the chemical vapor deposition of diamond films", Phys. Rev. B 1992, 46 1948]. In this paper, for the pure cupric element, the energy and structural parameters for several bulk phases as well as dimmer structure are well reproduced. The reference data are taken from our density functional theory calculations and the available experiments. The model potential also provides a good description of the bulk properties of various solid structures of cupric oxide compound structures, including cohesive energies, lattice parameters, and elastic constants. read less NOT USED (high confidence) K. Henriksson, C. Björkas, and K. Nordlund, “Atomistic simulations of stainless steels: a many-body potential for the Fe–Cr–C system,” Journal of Physics: Condensed Matter. 2013. link Times cited: 65 Abstract: Stainless steels found in real-world applications usually ha… read moreAbstract: Stainless steels found in real-world applications usually have some C content in the base Fe–Cr alloy, resulting in hard and dislocation-pinning carbides—Fe3C (cementite) and Cr23C6—being present in the finished steel product. The higher complexity of the steel microstructure has implications, for example, for the elastic properties and the evolution of defects such as Frenkel pairs and dislocations. This makes it necessary to re-evaluate the effects of basic radiation phenomena and not simply to rely on results obtained from purely metallic Fe–Cr alloys. In this report, an analytical interatomic potential parameterization in the Abell–Brenner–Tersoff form for the entire Fe–Cr–C system is presented to enable such calculations. The potential reproduces, for example, the lattice parameter(s), formation energies and elastic properties of the principal Fe and Cr carbides (Fe3C, Fe5C2, Fe7C3, Cr3C2, Cr7C3, Cr23C6), the Fe–Cr mixing energy curve, formation energies of simple C point defects in Fe and Cr, and the martensite lattice anisotropy, with fair to excellent agreement with empirical results. Tests of the predictive power of the potential show, for example, that Fe–Cr nanowires and bulk samples become elastically stiffer with increasing Cr and C concentrations. High-concentration nanowires also fracture at shorter relative elongations than wires made of pure Fe. Also, tests with Fe3C inclusions show that these act as obstacles for edge dislocations moving through otherwise pure Fe. read less NOT USED (high confidence) L. Pastewka, A. Klemenz, P. Gumbsch, and M. Moseler, “Screened empirical bond-order potentials for Si-C,” Physical Review B. 2013. link Times cited: 110 Abstract: Typical empirical bond-order potentials are short ranged and… read moreAbstract: Typical empirical bond-order potentials are short ranged and give ductile instead of brittle behavior for materials such as crystalline silicon or diamond. Screening functions can be used to increase the range of these potentials. We outline a general procedure to combine screening functions with bond-order potentials that does not require to refit any of the potential's properties. We use this approach to modify Tersoff's [Phys. Rev. B 39, 5566 (1989)], Erhart & Albe's [Phys. Rev. B 71, 35211 (2005)] and Kumagai et al.'s [Comp. Mater. Sci. 39, 457 (2007)] Si, C and Si-C potentials. The resulting potential formulations correctly reproduce brittle materials response, and give an improved description of amorphous phases. read less NOT USED (high confidence) M. Backman, N. Juslin, and K. Nordlund, “Bond order potential for gold,” The European Physical Journal B. 2012. link Times cited: 11 NOT USED (high confidence) L. Pastewka, M. Mrovec, M. Moseler, and P. Gumbsch, “Bond order potentials for fracture, wear, and plasticity,” MRS Bulletin. 2012. link Times cited: 55 Abstract: Coulson’s bond order is a chemically intuitive quantity that… read moreAbstract: Coulson’s bond order is a chemically intuitive quantity that measures the difference in the occupation of bonding and anti-bonding orbitals. Both empirical and rigorously derived bond order expressions have evolved in the course of time and proven very useful for atomistic modeling of materials. The latest generation of empirical formulations has recently been augmented by screening-function approaches. Using friction and wear of diamond and diamond-like carbon as examples, we demonstrate that such a screened bond order scheme allows for a faithful description of dynamical bond-breaking processes in materials far from equilibrium. The rigorous bond order expansions are obtained by systematic coarse-graining of the tight binding approximation and form a bridge between the electronic structure and the atomistic modeling hierarchies. They have enabled bottom-up derivations of bond order potentials for covalently bonded semiconductors, transition metals, and multicomponent intermetallics. The recently developed magnetic bond order potential gives a correct description of both directional covalent bonds and magnetic interactions in iron and is able to correctly predict the stability of bulk Fe polymorphs as well as the intricate properties of dislocation cores. The bond order schemes hence represent a family of reliable and powerful models that can be applied in large-scale simulations of complex processes involving fracture, wear, and plasticity. read less NOT USED (high confidence) S. Peng et al., “Bond-Order Potential for Erbium-Hydride System,” Journal of Physical Chemistry C. 2011. link Times cited: 7 Abstract: Interatomic potentials for an Er-H system are derived based … read moreAbstract: Interatomic potentials for an Er-H system are derived based on an analytical bond-order scheme. The model potentials provide a good description of the bulk properties and defect properties of hcp-Er, including lattice parameter, cohesive energy, elastic constants, point defect formation energies, surface and stacking fault energies. In addition to experimental data, a DFT method is used to construct the necessary database of different phases. We demonstrate that such potentials can reproduce the hydrogen behaviour in an alpha-phase Er-H system for a low hydrogen/metal ratio. Especially, the present potentials can be employed for modelling the energetics and structural properties of fcc ErH2, including lattice parameters, elastic constants, bulk modulus, Young's modulus and shear modulus, as well as the formation energies and migration barriers of point defects in ErH2. read less NOT USED (high confidence) M. Hu, K. Giapis, and D. Poulikakos, “Interfacial mixing during annealing of zinc oxide nanoparticle junctions,” Applied Physics Letters. 2011. link Times cited: 9 Abstract: The process of forming a junction between crystalline zinc o… read moreAbstract: The process of forming a junction between crystalline zinc oxide (ZnO) nanoparticles during pulsed thermal annealing in liquid tetradecane is studied using molecular dynamics simulation. Pairs of equal and unequal size particles are considered with emphasis on neck growth and atom mixing. The contact area and interface width of the junction are found to increase with heat pulse power albeit at different rates. The results suggest that it is possible to increase the junction area without significant mixing of atoms across the junction interface by tailoring the heat pulse power. read less NOT USED (high confidence) P. Kessler, K. Lorenz, and R. Vianden, “Implanted Impurities in Wide Band Gap Semiconductors,” Defect and Diffusion Forum. 2011. link Times cited: 8 Abstract: Wide band gap semiconductors, mainly GaN, have experienced m… read moreAbstract: Wide band gap semiconductors, mainly GaN, have experienced much attention due to their application in photonic devices and high-power or high-temperature electronic devices. Especially the synthesis of InxGa1-xN alloys has been studied extensively because of their use in LEDs and laser diodes. Here, In is added during the growth process and devices are already very successful on a commercial scale. Indium in nitride ternary and quaternary alloys plays a special role; however, the mechanisms leading to more efficient light emission in In-containing nitrides are still under debate. Therefore, the behaviour of In in GaN and AlN, the nitride semiconductor with the largest bandgap is an important field of study. In is also an important impurity in another wide band gap semiconductor – the II-VI compound ZnO where it acts as an n-type dopant. In this context the perturbed angular correlation technique using implantation of the probe 111In is a unique tool to study the immediate lattice environment of In in the wurtzite lattice of these wide band gap semiconductors. For the production of GaN and ZnO based electronic circuits one would normally apply the ion implantation technique, which is the most widely used method for selective area doping of semiconductors like Si and GaAs. However, this technique suffers from the fact that it invariably produces severe lattice damage in the implanted region, which in nitride semiconductors has been found to be very difficult to recover by annealing. The perturbed angular correlation technique is employed to monitor the damage recovery around implanted atoms and the properties of hitherto known impurity – defect complexes will be described and compared to proposed structure models. read less NOT USED (high confidence) A. Krasheninnikov and K. Nordlund, “Ion and electron irradiation-induced effects in nanostructured materials,” Journal of Applied Physics. 2010. link Times cited: 877 Abstract: A common misconception is that the irradiation of solids wit… read moreAbstract: A common misconception is that the irradiation of solids with energetic electrons and ions has exclusively detrimental effects on the properties of target materials. In addition to the well-known cases of doping of bulk semiconductors and ion beam nitriding of steels, recent experiments show that irradiation can also have beneficial effects on nanostructured systems. Electron or ion beams may serve as tools to synthesize nanoclusters and nanowires, change their morphology in a controllable manner, and tailor their mechanical, electronic, and even magnetic properties. Harnessing irradiation as a tool for modifying material properties at the nanoscale requires having the full microscopic picture of defect production and annealing in nanotargets. In this article, we review recent progress in the understanding of effects of irradiation on various zero-dimensional and one-dimensional nanoscale systems, such as semiconductor and metal nanoclusters and nanowires, nanotubes, and fullerenes. We also consider the t... read less NOT USED (high confidence) C. Sanz-Navarro et al., “Molecular Dynamics Simulations of Carbon-Supported Ni Clusters Using the Reax Reactive Force Field,” Journal of Physical Chemistry C. 2008. link Times cited: 30 Abstract: Molecular dynamics simulations have been performed using a R… read moreAbstract: Molecular dynamics simulations have been performed using a Reax force field for C/H/Ni systems to study the structural changes of an Ni_(100) cluster adsorbed on a carbon platelet. Three different edges of a carbon platelet are considered. We find a complete restructuring of the initial structure of the Ni_(100) clusters adsorbed on the armchair and zigzag edges. Nonetheless, the mean Ni−Ni bond length hardly changes. Several preferential sites on each of the graphite edges are identified. Diffusion of the entire cluster is found both for adsorption on the basal plane and for binding to a hydrogen terminated graphite edge. read less NOT USED (high confidence) D. Tainoff, B. Masenelli, O. Boisron, G. Guiraud, and P. Mélinon, “Crystallinity, Stoichiometry, and Luminescence of High Quality ZnO Nanoclusters,” Journal of Physical Chemistry C. 2008. link Times cited: 26 Abstract: Quasiperfect ZnO nanoclusters (∼6 nm in diameter) are synthe… read moreAbstract: Quasiperfect ZnO nanoclusters (∼6 nm in diameter) are synthesized in non steady state by an original process (the low energy cluster beam deposition, LECBD). The drastic cooling rate (10 10 K/s) of this cluster source induces an improvement of nanometric ZnO cluster quality as compared to more standard conditions of growth. The enhancement of the particle quality is justified by kinetics growth argument associated with X-ray photoelectron and Auger spectroscopies. Finally, we correlate the structural analysis to in situ cathodoluminescence spectroscopy. The study confirms the importance of the crystallinity more than the stoichiometry itself on the luminescence of the nanoparticles. read less NOT USED (high confidence) A. Caro, M. Caro, P. Klaver, B. Sadigh, E. M. Lopasso, and S. G. Srinivasan, “The computational modeling of alloys at the atomic scale: From ab initio and thermodynamics to radiation-induced heterogeneous precipitation,” JOM. 2007. link Times cited: 19 |
 Tersoff_LAMMPS_ErhartJuslinGoy_2006_ZnO__MO_616776018688_004
Tersoff_LAMMPS_ErhartJuslinGoy_2006_ZnO__MO_616776018688_004