Interatomic potential for Carbon (C), Hydrogen (H), Nitrogen (N), Oxygen (O). Use this Potential
Citing article:
Current potential: Sim_LAMMPS_ReaxFF_StrachanVanDuinChakraborty_2003_CHNO__SM_107643900657_001
Deep Citation determination:
Does the citing paper use the current potential to generate results displayed in the paper?
Provide us with identifying information so that we know you are not a bot (you will not be added to a mailing list):
Title
A single sentence description.
LAMMPS ReaxFF potential for RDX (C-H-N-O) systems developed by Strachan et al. (2003) v001
Description
LAMMPS ReaxFF potential for RDX (C-H-N-O) systems ('pair_style reaxff' with potential file ffield.reax.rdx and additional control and charge equilibration information). The parameters of the nitramine ReaxFF are based on a large number of ab initio QM calculations. Over 40 reactions and over 1600 equilibrated molecules have been used; they are designed to characterize the atomic interactions under various environments likely and unlikely high energy each atom can encounter. The training set contains bond breaking and compression curves for all possible bonds, angle and torsion bending data for all possible cases, as well as crystal data. See the supplemental material from Phys. Rev. Lett. 2003, 91, 098301 for a detailed description of the parameterization of this force field.
Species
The supported atomic species.
C, H, N, O
Disclaimer
A statement of applicability provided by the contributor, informing users of the intended use of this KIM Item.
This Simulator Model originally published in [1] is archived in OpenKIM [2-4].
[1] Strachan A, Duin ACT van, Chakraborty D, Dasgupta S, Goddard WA. Shock Waves in High-Energy Materials: The Initial Chemical Events in Nitramine RDX. Physical Review Letters. 2003Aug;91(9):098301. doi:10.1103/PhysRevLett.91.098301 — (Primary Source) A primary source is a reference directly related to the item documenting its development, as opposed to other sources that are provided as background information.
[2] Chakraborty D, Dasgupta S, Duin ACT van, Strachan A, Goddard WA. LAMMPS ReaxFF potential for RDX (C-H-N-O) systems developed by Strachan et al. (2003) v001. OpenKIM; 2020. doi:10.25950/ecee6dc8
[3] Tadmor EB, Elliott RS, Sethna JP, Miller RE, Becker CA. The potential of atomistic simulations and the Knowledgebase of Interatomic Models. JOM. 2011;63(7):17. doi:10.1007/s11837-011-0102-6
This panel presents information regarding the papers that have cited the interatomic potential (IP) whose page you are on.
The OpenKIM machine learning based Deep Citation framework is used to determine whether the citing article actually used the IP in computations (denoted by "USED") or only provides it as a background citation (denoted by "NOT USED"). For more details on Deep Citation and how to work with this panel, click the documentation link at the top of the panel.
The word cloud to the right is generated from the abstracts of IP principle source(s) (given below in "How to Cite") and the citing articles that were determined to have used the IP in order to provide users with a quick sense of the types of physical phenomena to which this IP is applied.
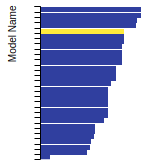
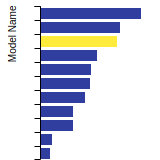
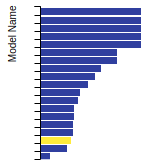
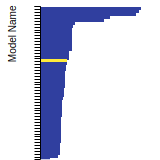
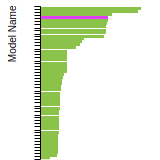
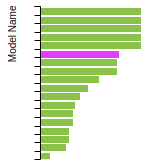
The bar chart shows the number of articles that cited the IP per year. Each bar is divided into green (articles that USED the IP) and blue (articles that did NOT USE the IP).
Users are encouraged to correct Deep Citation errors in determination by clicking the speech icon next to a citing article and providing updated information. This will be integrated into the next Deep Citation learning cycle, which occurs on a regular basis.
This panel provides information on past usage of this interatomic potential (IP) powered by the OpenKIM Deep Citation framework. The word cloud indicates typical applications of the potential. The bar chart shows citations per year of this IP (bars are divided into articles that used the IP (green) and those that did not (blue)). The complete list of articles that cited this IP is provided below along with the Deep Citation determination on usage. See the Deep Citation documentation for more information.
403 Citations (36 used)
Help us to determine which of the papers that cite this potential actually used it to perform calculations. If you know, click the .
USED (definite) S. Sun, F. Shan, Q. Lyu, C. Li, and S. Hu, “Theoretical Prediction of Mechanical Strength and Desalination Performance of One-Atom-Thick Hydrocarbon Polymer in Pressure-Driven Separation,” Polymers. 2019. link Times cited: 2
Abstract: One-atom-thick materials hold promise for the future of memb… read more
Abstract: One-atom-thick materials hold promise for the future of membrane-based gas purification and water filtration applications. However, there are a few investigations on the mechanical properties of these materials under pressure-driven condition. Here, by employing molecular simulation techniques and continuum mechanics simulation, we investigate the mechanical strength of two-dimensional hydrocarbon polymers containing sub-nanometer pores with various topologies. We demonstrate that the mechanical strengths of the membranes are correlated with their pore sizes and geometries. In addition, when the pore size of substrates is controlled within a reasonable range, all of the membrane candidates can withstand the practical hydraulic pressure of few megapascal. The studied materials also exhibit better seawater desalination performance as compared to the traditional polymeric reverse osmosis membrane. This work presents a new route to design new separation membrane, and also propose a simulation method to evaluate the mechanical strength and desalination performance. read less
USED (definite) P. Vashishta, R. Kalia, A. Nakano, B. Homan, and K. McNesby, “Multimillion Atom Reactive Simulations of Nanostructured Energetic Materials,” Journal of Propulsion and Power. 2007. link Times cited: 14
Abstract: DOI: 10.2514/1.25651For large-scale atomistic simulations in… read more
Abstract: DOI: 10.2514/1.25651For large-scale atomistic simulations involving chemical reactions to study nanostructured energetic materials, wehave designed linear-scaling molecular dynamics algorithms: 1) ! rst-principles-based fast reactive force ! eldmolecular dynamics, and 2) embedded divide-and-conquer density functional theory on adaptive multigrids forquantum-mechanical molecular dynamics. These algorithms have achieved unprecedented scales of quantum-mechanically accurate and well validated, chemically reactive atomistic simulations [0.56 billion-atom ! rstprinciples-based fast reactive force ! eld molecular dynamics and 1.4 million-atom (0.12 trillion grid points)embedded divide-and-conquer density functional theory molecular dynamics] in addition to 18.9 billion-atomnonreactive space-time multiresolution molecular dynamics, with parallel ef! ciency as high as 0.953 on 1920Itanium2 processors. These algorithms have enabled us to perform reactive molecular dynamics simulations toreveal various atomistic processes during 1) the oxidation of an aluminum nanoparticle, 2) the decomposition andchemisorption of an RDX (1, 3, 5-trinitro-1, 3, 5-triazine) molecule on an aluminum surface, and 3) shock-initiateddetonation of energetic nanocomposite material (RDX crystalline matrix embedded with aluminum nanoparticles. read less
USED (high confidence) O. Sergeev, A. Mukhanov, S. Murzov, and A. Yanilkin, “Complete equations of state for PETN and its products from atomistic simulations.,” Physical chemistry chemical physics : PCCP. 2020. link Times cited: 2
Abstract: The complete caloric and thermal equations of state for pent… read more
Abstract: The complete caloric and thermal equations of state for pentaerythritol tetranitrate (PETN) and its decomposition products are developed. The equation for the crystalline state is obtained with quasiharmonic approximation for the vibrational energy, with the force constants being calculated using density functional theory. The equation of state for the products is derived from equilibrium ReaxFF molecular dynamics simulations. Two equations are coupled through the heat of thermal decomposition calculated using ReaxFF at high temperature. Our hydrodynamic code utilizing the developed EOSs reproduces well the detonation velocity and Chapman-Jouguet pressure obtained in the molecular dynamics simulations. read less
USED (high confidence) X. Huang et al., “Anisotropic hydrogen bond structures and orientation dependence of shock sensitivity in crystalline 1,3,5-tri-amino-2,4,6-tri-nitrobenzene (TATB).,” Physical chemistry chemical physics : PCCP. 2020. link Times cited: 9
Abstract: The orientation dependence of shock sensitivity in high expl… read more
Abstract: The orientation dependence of shock sensitivity in high explosive crystals was explored in this study. As a widely used wood explosive, 1,3,5-tri-amino-2,4,6-tri-nitrobenzene (TATB) is insensitive to thermal ignition and mechanical impact. Its typical anisotropic crystal structure suggests anisotropic shock sensitivity. Shockwaves were applied to an incised TATB crystal along three orthogonal directions using the multiscale shock technique (MSST) combined with the ReaxFF method to study the origin of anisotropic shock sensitivity. The physical and chemical responses of the TATB crystal during shock were investigated. The results show that the temperature, stress, volume compressibility, and decomposition rate of TATB are strongly dependent on the shockwave direction. In other words, the sensitivity of TATB to mechanical shock is strongly dependent on the crystal orientation. TATB is relatively sensitive along the directions parallel to the (001) crystal plane (X and Y directions) and is highly insensitive along the [001] direction (Z direction). We calculated the energy of intermolecular hydrogen bonds and the elastic constants of the TATB crystal using ab initio simulations, which also show anisotropy. We found that the unique structure of intermolecular hydrogen bonds and the difference in temperature rise induced by orientation-related compressibility are primarily responsible for the anisotropic shock wave sensitivity. read less
USED (high confidence) J. Li et al., “Reactive molecular dynamics simulations on the thermal decompositions and oxidations of TKX-50 and twinned TKX-50,” CrystEngComm. 2020. link Times cited: 16
Abstract: The influence of twinned crystals on the performance of TKX-… read more
Abstract: The influence of twinned crystals on the performance of TKX-50 is investigated using normal TKX-50 and twinned TKX-50 supercells. ReaxFF-lg reactive molecular dynamics simulations are performed to study thermal decomposition and oxidation. read less
USED (high confidence) M. Shishehbor and M. Pouranian, “Tuning the Mechanical and Adhesion Properties of Carbon Nanotubes Using Aligned Cellulose Wrap (Cellulose Nanotube): A Molecular Dynamics Study,” Nanomaterials. 2020. link Times cited: 11
Abstract: Improving the adhesion properties of carbon nanotubes (CNTs)… read more
Abstract: Improving the adhesion properties of carbon nanotubes (CNTs) at the molecular scale can significantly enhance dispersion of CNT fibers in polymer matrix and unleash the dormant extraordinary mechanical properties of CNTs in CNT-polymer nanocomposites. Inspired by the outstanding adhesion, dispersion, mechanical, and surface functionalization properties of crystalline nanocellulose (CNC), this paper studies the mechanical and adhesion properties of CNT wrapped by aligned cellulose chains around CNT using molecular dynamic simulations. The strength, elastic modulus, and toughness of CNT-cellulose fiber for different cellulose contents are obtained from tensile and compression tests. Additionally, the effect of adding cellulose on the surface energy, interfacial shear modulus, and strength is evaluated. The result shows that even adding a single layer cellulose wrap (≈55% content) significantly decreases the mechanical properties, however, it also dramatically enhances the adhesion energy, interfacial shear strength, and modulus. Adding more cellulose layers, subsequently, deceases and increases mechanical properties and adhesion properties, respectively. In addition, analysis of nanopapers of pristine CNT, pristine CNC, and CNT-wrapped cellulose reveals that CNT-wrapped cellulose nanopapers are strong, stiff, and tough, while for CNT and CNC either strength or toughness is compromised. This research shows that cellulose wraps provide CNT fibers with tunable mechanical properties and adhesion energy that could yield strong and tough materials due to the excellent mechanical properties of CNT and active surface and hydrogen bonding of cellulose. read less
USED (high confidence) I. Leven and T. Head‐Gordon, “Inertial extended-Lagrangian scheme for solving charge equilibration models.,” Physical chemistry chemical physics : PCCP. 2019. link Times cited: 11
Abstract: The inertial extended Lagrangian/self-consistent field schem… read more
Abstract: The inertial extended Lagrangian/self-consistent field scheme (iEL-SCF) has been adopted for solving charge equilibration in LAMMPS as part of the reactive force field ReaxFF, which due to the charge conservation constraint requires solving two sets of linear system of equations for the new charges at each molecular dynamics time-step. Therefore, the extended Lagrangian for charge equilibration is comprised of two auxiliary variables for the intermediate charges which serve as an initial guess for the real charges. We show that the iEL-SCF is able to reduce the number of SCF cycles by 50-80% of the original conjugate gradient self-consistent field solver as tested across diverse systems including water, ferric hydroxide, nitramine RDX, and hexanitrostilbene. read less
USED (high confidence) J. M. Sousa et al., “Elastic properties of graphyne-based nanotubes,” Computational Materials Science. 2019. link Times cited: 30
USED (high confidence) Y. Thakur et al., “Generating high dielectric constant blends from lower dielectric constant dipolar polymers using nanostructure engineering,” Nano Energy. 2017. link Times cited: 77
USED (high confidence) C. Zhang et al., “Sequential Molecular Dynamics Simulations: A Strategy for Complex Chemical Reactions and a Case Study on the Graphitization of Cooked 1,3,5-Triamino-2,4,6-trinitrobenzene,” Journal of Physical Chemistry C. 2016. link Times cited: 20
Abstract: The fundamental core of chemistry is to create new substance… read more
Abstract: The fundamental core of chemistry is to create new substances, and numerous complex reactions may be involved in chemical conversions. Nevertheless, clarifying the mechanisms of these complex reactions remains challenging, thereby causing insufficiencies in the fundamentals to guide new substance creation. This work proposes and emphasizes a strategy of sequential molecular dynamics simulations (SMDSs) toward complex chemical reactions. The strategy is successfully demonstrated by clarifying a complex graphitization process of 1,3,5-triamino-2,4,6-trinitrobenzene (TATB), whose mechanism has not been imaged by a single simulation alone. We conducted SMDSs with a molecular reactive force field, ReaxFF, to resemble the cook-off of TATB, i.e., a sequence of heating, expansion, and cooling acting on TATB. Graphitization is found to sequentially undergo TATB molecular decay, clustering, cluster enlargement to C sheets (sheeting), and layered stacking of C sheets, along with phase separation. Moreover, the struc... read less
USED (high confidence) Y. Yao et al., “Carbon Welding by Ultrafast Joule Heating.,” Nano letters. 2016. link Times cited: 74
Abstract: Carbon nanomaterials exhibit outstanding electrical and mech… read more
Abstract: Carbon nanomaterials exhibit outstanding electrical and mechanical properties, but these superior properties are often compromised as nanomaterials are assembled into bulk structures. This issue of scaling limits the use of carbon nanostructures and can be attributed to poor physical contacts between nanostructures. To address this challenge, we propose a novel technique to build a 3D interconnected carbon matrix by forming covalent bonds between carbon nanostructures. High temperature Joule heating was applied to bring the carbon nanofiber (CNF) film to temperatures greater than 2500 K at a heating rate of 200 K/min to fuse together adjacent carbon nanofibers with graphitic carbon bonds, forming a 3D continuous carbon network. The bulk electrical conductivity of the carbon matrix increased four orders of magnitude to 380 S/cm with a sheet resistance of 1.75 Ω/sq. The high temperature Joule heating not only enables fast graphitization of carbon materials at high temperature, but also provides a new strategy to build covalently bonded graphitic carbon networks from amorphous carbon source. Because of the high electrical conductivity, good mechanical structures, and anticorrosion properties, the 3D interconnected carbon membrane shows promising applications in energy storage and electrocatalysis fields. read less
USED (high confidence) N. Wang, J.-hua Peng, A.-min Pang, J. Hu, and T. He, “Study on the anisotropic response of condensed-phase RDX under repeated stress wave loading via ReaxFF molecular dynamics simulation,” Journal of Molecular Modeling. 2016. link Times cited: 4
USED (high confidence) Y. Wen, X. Xue, X. Long, and C. Zhang, “Cluster Evolution at Early Stages of 1,3,5-Triamino-2,4,6-trinitrobenzene under Various Heating Conditions: A Molecular Reactive Force Field Study.,” The journal of physical chemistry. A. 2016. link Times cited: 49
Abstract: We carried out reactive molecular dynamics simulations by Re… read more
Abstract: We carried out reactive molecular dynamics simulations by ReaxFF to study the initial events of an insensitive high explosive 1,3,5-triamino-2,4,6-trinitrobenzene (TATB) against various thermal stimuli including constant-temperature heating, programmed heating, and adiabatic heating to simulate TATB suffering from accidental heating in reality. Cluster evolution at the early stage of the thermal decomposition of condensed TATB was the main focus as cluster formation primarily occurs when TATB is heated. The results show that cluster formation is the balance of the competition of intermolecular collision and molecular decomposition of TATB, that is, an appropriate temperature and certain duration are required for cluster formation and preservation. The temperature in the range of 2000-3000 K was found to be optimum for fast formation and a period of preservation. Besides, the intra- and intermolecular H transfers are always favorable, whereas the C-NO2 partition was favorable at high temperature. The simulation results are helpful to deepen the insight into the thermal properties of condensed TATB. read less
USED (high confidence) R. Dong, V. Ranjan, M. Nardelli, and J. Bernholc, “Atomistic simulations of aromatic polyurea and polyamide for capacitive energy storage,” Physical Review B. 2015. link Times cited: 10
Abstract: Materials for capacitive energy storage with high energy den… read more
Abstract: Materials for capacitive energy storage with high energy density and low loss are desired in many fields. We investigate several polymers with urea and amide functional groups using density functional theory and classical molecular dynamics simulations. For aromatic polyurea (APU) and para-aramid (PA), we find several nearly energetically degenerate ordered structures, while meta-aromatic polyurea (mAPU) tends to be rotationally disordered along the polymer chains. Simulated annealing of APU and PA structures results in the formation of hydrogen-bonded sheets, highlighting the importance of dipole-dipole interactions. In contrast, hydrogen bonding does not play a significant role in mAPU, hence the propensity to disorder. We find that the disordered structures with misaligned chains have significantly larger dielectric constants, due to significant increase in the free volume, which leads to easier reorientation of dipolar groups in the presence of an electric field. Large segment motion is still not allowed below the glass transition temperature, which explains the experimentally observed very low loss at high field and elevated temperature. However, the degree of disorder needs to be controlled, because highly entangled structures diminish the free dipoles and decrease permittivity. Among the considered materials, mAPU is the most promising dielectric for capacitive energy storage, but the concept of increasing permittivity while maintaining low loss through disorder-induced free volume increase is generally applicable and provides an alternative pathway for the design of high-performance dielectrics for capacitive energy storage. read less
USED (high confidence) M. Warrier, P. Pahari, and S. Chaturvedi, “Molecular dynamics analysis of the transient temperature increase at void locations in shocked materials: RDX and Cu,” Journal of Molecular Modeling. 2015. link Times cited: 5
USED (high confidence) S. Park et al., “In situ hybridization of carbon nanotubes with bacterial cellulose for three-dimensional hybrid bioscaffolds.,” Biomaterials. 2015. link Times cited: 75
USED (high confidence) X. Dong, X. Fan, Y. Fan, and Y. Wen, “Reactive molecular dynamics simulation of the pyrolysis and combustion of benzene: ultrahigh temperature and oxygen-induced enhancement of initiation pathways and their effect on carbon black generation,” RSC Advances. 2015. link Times cited: 12
Abstract: The pyrolysis and combustion mechanisms of benzene under dif… read more
Abstract: The pyrolysis and combustion mechanisms of benzene under different chemical environments and temperatures were investigated by a reactive force field based molecular dynamics (ReaxFF MD) simulation using two systems, pure benzene and a mixture of benzene and oxygen gas. The chemical behaviors of this system were investigated under an ultrahigh temperature that can be induced by a high-energy density laser and compared to those at high temperature. According to some experimental data, we assume that an ultrahigh temperature can be used to mimic laser irradiation. The conclusions of this simulation are as follows. First, the ReaxFF MD simulations showed that the decomposition rates of benzene were significantly accelerated by laser irradiation or in the presence of oxygen. Second, additional initiation pathways were opened up by these two factors. The primary initiation pathway involves only the hydrogen atom loss in the pyrolysis of benzene at 3000 K, and the initiation pathways become much more complicated after laser irradiation or the involvement of oxygen. Third, the ReaxFF MD simulations formed a reasonable carbon black (CB) texture of various sizes in the pyrolysis of benzene, and we also focused on the evolution of the texture of CB. The calculation results of the final gaseous products, hydrocarbons, and the formation of CB are in a good agreement with the literature. This study provides a better understanding of the initiation mechanisms of the pyrolysis and combustion of benzene under extreme conditions. read less
USED (high confidence) J.-Y. Li et al., “Ab initio molecular dynamics simulation on the formation process of He@C60 synthesized by explosion,” Journal of Molecular Modeling. 2013. link Times cited: 2
USED (high confidence) E. Reed et al., “A new mechanism for observation of THz acoustic waves: coherent THz radiation emission,” OPTO. 2009. link Times cited: 0
Abstract: Our simulations and experiments demonstrate a new physical m… read more
Abstract: Our simulations and experiments demonstrate a new physical mechanism for detecting acoustic waves of THz frequencies. We find that strain waves of THz frequencies can coherently generate radiation when they propagate past an interface between materials with different piezoelectric coefficients. By considering AlN/GaN heterostructures, we show that the radiation is of detectable amplitude and contains sufficient information to determine the time-dependence of the strain wave with potentially sub-picosecond, nearly atomic time and space resolution. This mechanism is distinct from optical approaches to strain wave measurement. We demonstrate this phenomenon within the context of high amplitude THz frequency strain waves that spontaneously form at the front of shock waves in GaN crystals. We also show how the mechanism can be utilized to determine the layer thicknesses in thin film GaN/AlN heterostructures. read less
USED (high confidence) S. S. Choi and C. E. Son, “Testing Method for On‐Site Measurement of Explosive Materials Contaminated on Travel Luggage Bag and Backpack Using Ion Mobility Spectrometry,” Bulletin of The Korean Chemical Society. 2018. link Times cited: 1
USED (high confidence) S. Cho, “Chemical Stability of Carbon Nanotube Containers Loaded with Nitromethane: Reactive Molecular Dynamic Simulation,” Bulletin of The Korean Chemical Society. 2017. link Times cited: 1
USED (low confidence) Y. Zhang, T. Wang, and Y. He, “Initial Response of Pentaerythritol Tetranitrate (PETN) under the Coupling Effect of Preheating, Shock and Defect via the Molecular Dynamics Simulations with the Multiscale Shock Technique Method,” Molecules. 2023. link Times cited: 0
Abstract: The initial response of PETN under the coupling of preheatin… read more
Abstract: The initial response of PETN under the coupling of preheating, impact and defects was simulated by Multiscale Shock Technique (MSST) method and molecular dynamics. The temperature change of PETN during impact compression can be divided into three stages: (1) the elastoplastic change of the system caused by initial compression; (2) part of PETN decomposes and releases energy to raise temperature; (3) a secondary chemical reaction occurs, resulting in rapid temperature rise. Under the given conditions, a higher initial preheating temperature will lead to faster decomposition of PETN; The existence of defects will accelerate the decomposition of PETN molecules; Coupling the highest preheating temperature with defects will lead to the fastest decomposition of PETN molecules, while in the defect-free PETN system with a preheating temperature of 300 K, the decomposition of PETN molecules is the slowest. For the case of Us = 8 km·s−1, the effect of defects on the initial PETN reaction is greater than the initial preheating temperature; When the impact velocity is greater than 9 km·s−1, the impact velocity is an important factor affecting the decomposition of PETN molecules. For Us = 10 km·s−1, NO2 is the main initial product in the defective PETN crystal, while in the perfect PETN crystal, it is the combination of NO2 and HONO. The chemical reaction kinetics analysis shows that the preheating temperature and defects will accelerate the decomposition of PETN. The higher the preheating temperature, the faster the decomposition of PETN. For the case of Us = 7 km·s−1, 8 km·s−1 and 9 km·s−1, the existence of defects will increase the decomposition rate by more than 50% regardless of the initial preheating temperature. In the case of Us = 10 km·s−1, the improvement of decomposition rate by defects is not as significant as the initial preheating temperature. read less
USED (low confidence) P. Pahari, A. Rao, and M. Warrier, “Molecular dynamics simulations of the decomposition and U_s–U_p relationship of RDX molecular crystal subjected to high velocity impact,” Journal of Molecular Modeling. 2023. link Times cited: 0
USED (low confidence) M. Zhou, J. Luo, and D. Xiang, “Effects of Different Guests on Pyrolysis Mechanism of α-CL−20/Guest at High Temperatures by Reactive Molecular Dynamics Simulations at High Temperatures,” International Journal of Molecular Sciences. 2023. link Times cited: 0
Abstract: The host–guest inclusion strategy has the potential to surpa… read more
Abstract: The host–guest inclusion strategy has the potential to surpass the limitations of energy density and suboptimal performances of single explosives. The guest molecules can not only enhance the detonation performance of host explosives but also can enhance their stability. Therefore, a deep analysis of the role of guest influence on the pyrolysis decomposition of the host–guest explosive is necessary. The whole decomposition reaction stage of CL-20/H2O, CL-20/CO2, CL-20/N2O, CL-20/NH2OH was calculated by ReaxFF-MD. The incorporation of CO2, N2O and NH2OH significantly increase the energy levels of CL-20. However, different guests have little influence on the initial decomposition paths of CL-20. The Ea1 and Ea2 values of CL-20/CO2, CL-20/N2O, CL-20/NH2OH systems are higher than the CL-20/H2O system. Clearly, incorporation of CO2, N2O, NH2OH can inhibit the initial decomposition and intermediate decomposition stage of CL-20/H2O. Guest molecules become heavily involved in the reaction and influence on the reaction rates. k1 of CL-20/N2O and CL-20/NH2OH systems are significantly larger than that of CL-20/H2O at high temperatures. k1 of CL-20/CO2 system is very complex, which can be affected deeply by temperatures. k2 of the CL-20/CO2, CL-20/N2O systems is significantly smaller than that of CL-20/H2O at high temperatures. k2 of CL-20/NH2OH system shows little difference at high temperatures. For the CL-20/CO2 system, the k3 value of CO2 is slightly higher than that for CL-20/H2O, CL-20/N2O, CL-20/NH2OH systems, while the k3 values of N2 and H2O are slightly smaller than that for the CL-20/H2O, CL-20/N2O, CL-20/NH2OH systems. For the CL-20/N2O system, the k3 value of CO2 is slightly smaller than that for CL-20/H2O, CL-20/CO2, CL-20/NH2OH systems. For the CL-20/NH2OH system, the k3 value of H2O is slightly larger than that for CL-20/H2O, CL-20/CO2, CL-20/N2O systems. These mechanisms revealed that CO2, N2O and NH2OH molecules inhibit the early stages of the initial decomposition of CL-20 and play an important role for the decomposition subsequently. read less
USED (low confidence) Y. Zhang et al., “The Effects of Atomic Oxygen and Ion Irradiation Degradation on Multi-Polymers: A Combined Ground-based Exposure and ReaxFF-MD Simulation,” Polymer Degradation and Stability. 2022. link Times cited: 7
USED (low confidence) X. Li, L. Liu, H. Mei, S. Xu, J. Li, and J. Zhang, “The formation mechanisms of amorphous bands of boron carbide nanopillars under uniaxial compressions and their effects on mechanical properties from molecular dynamics simulation,” Computational Materials Science. 2021. link Times cited: 1
USED (low confidence) D. Hu, X. Gu, and B. Cui, “Effect of styrene-butadiene-styrene copolymer on the aging resistance of asphalt: An atomistic understanding from reactive molecular dynamics simulations,” Frontiers of Structural and Civil Engineering. 2021. link Times cited: 6
USED (low confidence) M. I. R. Shishir, M. S. Elapolu, and A. Tabarraei, “Investigation of fracture and mechanical properties of monolayer C3N using molecular dynamic simulations,” Mechanics of Materials. 2021. link Times cited: 6
USED (low confidence) G. Li et al., “Simulation of pyrolysis of crosslinked epoxy resin using ReaxFF molecular dynamics,” Computational and Theoretical Chemistry. 2021. link Times cited: 13
USED (low confidence) T. Le, A. Striolo, and D. Cole, “Partial CO2 Reduction in Amorphous Cylindrical Silica Nanopores Studied with Reactive Molecular Dynamics Simulations,” The Journal of Physical Chemistry C. 2019. link Times cited: 4
Abstract: It is known that pore confinement affects the structure and … read more
Abstract: It is known that pore confinement affects the structure and transport properties of fluids. It has also been shown that confinement can affect the equilibrium composition of a reactive system. Such effects could be related to the possible abiotic hydrocarbon synthesis in deep-sea hydrothermal vents, especially when the CO2 methanation reaction occurs within nanopores. In an attempt to identify possible rate-limiting steps of such a reaction, we report here molecular dynamics simulations conducted implementing the reactive force field. The reaction is considered within a cylindrical nanopore carved out of amorphous silica. Within the constraints of our simulations, which were conducted for 5 ns, no CH4 molecules were detected in the temperature range of 400–1000 K, suggesting that the silica pore hinders the complete CO2 reduction. This is consistent with the fact that silica is not an effective catalyst for CO2 methanation. Our simulations, in agreement with literature reports, suggest that the silica por... read less
USED (low confidence) F. Wang, L. Chen, D. Geng, J. Lu, and J. Wu, “Molecular Dynamics Simulations of an Initial Chemical Reaction Mechanism of Shocked CL-20 Crystals Containing Nanovoids,” The Journal of Physical Chemistry C. 2019. link Times cited: 18
Abstract: To understand the initial chemical reaction mechanism of the… read more
Abstract: To understand the initial chemical reaction mechanism of the heterogeneous explosive hexanitrohexaazaisowurtzitane (CL-20), it is necessary to study the shock initiation mechanism of this nanovoid-containing crystal. In this paper, supercells of CL-20 with different void sizes were constructed. The chemical reactions induced by different impact velocities were calculated using molecular dynamics based on the ReaxFF-lg reactive force field. The effects of impact velocities and void sizes on the chemical reactions of the CL-20 crystal were discussed. The initial reaction of CL-20 molecules around the voids was analyzed, and the evolution of the formation and breakage of chemical bonds as well as the elementary reactions were also obtained. It is found that under an impact, the CL-20 molecules around the voids first undergo polymerization of the N–O bonds and then breakage of the C–N, N–N, and C–H bonds occurs. Increased void size and impact velocity lead to higher temperature “hot spots” and more intense ch... read less
USED (low confidence) Y. Dong, M. Meng, M. M. Groves, C. Zhang, and J. Lin, “Thermal conductivities of two-dimensional graphitic carbon nitrides by molecule dynamics simulation,” International Journal of Heat and Mass Transfer. 2018. link Times cited: 50
USED (low confidence) V. Dozhdikov, A. Basharin, P. Levashov, and D. Minakov, “Atomistic simulations of the equation of state and hybridization of liquid carbon at a temperature of 6000 K in the pressure range of 1-25 GPa.,” The Journal of chemical physics. 2017. link Times cited: 16
Abstract: The equation of state and the structure of liquid carbon are… read more
Abstract: The equation of state and the structure of liquid carbon are studied by molecular simulation. Both classical and quantum molecular dynamics (QMD) are used to calculate the equation of state and the distribution of chemical bonds at 6000 K in the pressure range 1-25 GPa. Our calculations and results of other authors show that liquid carbon has a fairly low density on the order of 1.2-1.35 g/cm3 at pressures about 1 GPa. Owing to the coordination number analysis, this fact can be attributed to the high content of sp1-bonded atoms (more than 50% according to our ab initio computations). Six empirical potentials have been tested in order to describe the density dependence of pressure and structure at 6000 K. As a result, only one potential, ReaxFF/lg, was able to reproduce the QMD simulations for both the equation of state and the fraction of sp1, sp2, sp3-bonded atoms. read less
USED (low confidence) X.-P. Liu, J.-H. Zhan, D. Lai, X. Liu, Z. Zhang, and G. Xu, “Initial Pyrolysis Mechanism of Oil Shale Kerogen with Reactive Molecular Dynamics Simulation,” Energy & Fuels. 2015. link Times cited: 60
Abstract: Molecular dynamics (MD) simulations using a reactive force f… read more
Abstract: Molecular dynamics (MD) simulations using a reactive force field (ReaxFF) method for a Green River oil shale model demonstrate that the thermal decomposition of the oil shale molecule is initiated with the cleavage of the oxygen bridge (C-O bond), and the first product is formaldehyde (CH2O). The simulation results show that the C-O bond is weaker than the other bonds, agreeing with its smaller bond dissociation energy (BDE). The ring-opening position of the aliphatic ring is usually determined by the stability of free radicals formed in this process. For aromatic hydrocarbons, the long-chain substituents are found to be easier to leave and the cleavage of C-C bonds leads to a series of chain reactions and the formation of small fragments, such as ethylene and propylene. The bond cleavages are almost in accordance with the minimum bonding energy rule. NVT simulations show that the pyrolysis process progresses in two stages: the decomposition of kerogen into heavy (C40+.) species and then the generation of light compounds. Recombinations and rearrangements of different fragments are also observed via MD simulations. The main hydrocarbon fragments of C-10-C-20 are regarded as the component or precursor of diesel oil. The formation pathways of typical aromatic components are analyzed by tracking the motion trajectories of relevant structures. The intermediates and products in MD simulations are found to be similar to the gas chromatography mass spectrometry (GC-MS) results from previous experiments. read less
USED (low confidence) B. Chen, Z.-J. Diao, and H. Lu, “Using the ReaxFF reactive force field for molecular dynamics simulations of the spontaneous combustion of lignite with the Hatcher lignite model,” Fuel. 2014. link Times cited: 61
USED (low confidence) A. Shekhar, K. Nomura, R. Kalia, A. Nakano, and P. Vashishta, “Nanobubble collapse on a silica surface in water: billion-atom reactive molecular dynamics simulations.,” Physical review letters. 2013. link Times cited: 50
Abstract: Cavitation bubbles occur in fluids subjected to rapid change… read more
Abstract: Cavitation bubbles occur in fluids subjected to rapid changes in pressure. We use billion-atom reactive molecular dynamics simulations on a 163,840-processor BlueGene/P supercomputer to investigate damage caused by shock-induced collapse of nanobubbles in water near an amorphous silica surface. Collapse of an empty bubble generates a high-speed nanojet, which causes pitting on the silica surface. We find pit radii are close to bubble radii, and experiments also indicate linear scaling between them. The gas-filled bubbles undergo partial collapse and, consequently, the damage on the silica surface is mitigated. read less
NOT USED (low confidence) Y. Guo, H. Shi, H. Liu, Y. Xie, and Y. Guan, “Reactive molecular dynamics simulation and chemical kinetic modeling of ammonia/methane co-combustion,” Fuel. 2023. link Times cited: 0
NOT USED (low confidence) F. Xiong et al., “Mechanistic study of the influence of aluminum nanoparticles on the pressure sensitivity of 1,3,5-trinitro1,3,5-triazinane (RDX) thermal decomposition,” Colloids and Surfaces A: Physicochemical and Engineering Aspects. 2023. link Times cited: 0
NOT USED (low confidence) Z.-J. Sun, H. Li, and W. Zhu, “Reactive molecular dynamics simulations on the decomposition process of 1,3,5-trinitro-1,3,5-triazine crystal under high temperatures and pressure,” Journal of Molecular Modeling. 2023. link Times cited: 0
NOT USED (low confidence) H. Liu, C. She, C.-X. Yang, Z. Jin, X. Tan, and K. Chen, “A combined ReaxFF simulation and TG-MS study on the thermal decomposition mechanism of 5,5ʹ-dinitramino-3,3ʹ-bi[1,2,4-triazolate] carbohydrazide salt (CBNT),” Journal of Thermal Analysis and Calorimetry. 2023. link Times cited: 0
NOT USED (low confidence) U. Nwankwo, Y.-D. Wang, C. Lam, and N. Onofrio, “Charge equilibration model with shielded long-range Coulomb for reactive molecular dynamics simulations.,” The Journal of chemical physics. 2023. link Times cited: 1
Abstract: Atomic description of electrochemical systems requires react… read more
Abstract: Atomic description of electrochemical systems requires reactive interaction potential to explicitly describe the chemistry between atoms and molecules and the evolving charge distribution and polarization effects. Calculating Coulomb electrostatic interactions and polarization effects requires a better estimate of the partial charge distribution in molecular systems. However, models such as reactive force fields and charge equilibration (QEq) include Coulomb interactions up to a short-distance cutoff for better computational speeds. Ignoring long-distance electrostatic interaction affects the ability to describe electrochemistry in large systems. We studied the long-range Coulomb effects among charged particles and extended the QEq method to include long-range effects. By this extension, we anticipate a proper account of Coulomb interactions in reactive molecular dynamics simulations. We validate the approach by computing charges on a series of metal-organic frameworks and some simple systems. Results are compared to regular QEq and quantum mechanics calculations. The study shows slightly overestimated charge values in the regular QEq approach. Moreover, our method was combined with Ewald summation to compute forces and evaluate the long-range effects of simple capacitor configurations. There were noticeable differences between the calculated charges with/without long-range Coulomb interactions. The difference, which may have originated from the long-range influence on the capacitor ions, makes the Ewald method a better descriptor of Coulomb electrostatics for charged electrodes. The approach explored in this study enabled the atomic description of electrochemical systems with realistic electrolyte thickness while accounting for the electrostatic effects of charged electrodes throughout the dielectric layer in devices like batteries and emerging solid-state memory. read less
NOT USED (low confidence) R. Cappabianca, P. D. Angelis, M. Fasano, E. Chiavazzo, and P. Asinari, “An Overview on Transport Phenomena within Solid Electrolyte Interphase and Their Impact on the Performance and Durability of Lithium-Ion Batteries,” Energies. 2023. link Times cited: 0
Abstract: The nature of the electrode–electrolyte interface has an imp… read more
Abstract: The nature of the electrode–electrolyte interface has an impact on the performance and durability of lithium-ion batteries (LIBs). The initial electrolyte’s thermodynamic instability at the anode–electrolyte interface in LIBs results in the formation of a passivation layer, called solid electrolyte interphase (SEI). The initial dense and intact layer allows Li+ transport and restricts electron tunneling, thus preventing electrolyte decomposition and ensuring the electrochemical stability of a battery. However, the growth of this layer can reduce the availability of active lithium and electrolyte, and ultimately lead to an irreversible battery capacity fade. Investigating the transport phenomena of lithium ions within SEI is crucial for understanding its formation and growth. Nonetheless, accurately describing all relevant mechanisms is challenging due to its complex and multiscale nature. An overview of current computational efforts to study Li+ transport within SEI is given in this article, ranging from electronic/atomistic scale simulations to macroscopic models. The drawbacks and advantages of the proposed numerical approaches are summarized along with the obstacles that need to be overcome to obtain accurate experimental data, identified on the basis of the most recent literature evidence. We highlight collaboration gaps between modeling and experimental approaches, as well as the urgent need for new multiscale models, to gain a better understanding of such a crucial transport phenomenon. read less
NOT USED (low confidence) Y. Sha and X. Zhang, “Reaction mechanism of hydrogen peroxide enhancing detonation performance in the host-guest structure of CL-20 by reactive molecular dynamics simulations,” Vacuum. 2023. link Times cited: 0
NOT USED (low confidence) R. Zhang et al., “Study on pyrolysis mechanism of 1,7-diacetoxy-2,4,6-trinitro-2,4,6-triazaheptane (BSX),” Arabian Journal of Chemistry. 2023. link Times cited: 0
NOT USED (low confidence) B. Hamilton and T. Germann, “Using limited neural networks to assess relative mechanistic influence on shock heating in granular solids,” Physical Review Materials. 2023. link Times cited: 0
Abstract: The rapid compaction of granular media results in localized … read more
Abstract: The rapid compaction of granular media results in localized heating that can induce chemical reactions, phase transformations, and melting. However, there are numerous mechanisms in play that can be dependent on a variety of microstructural features. Machine learning techniques such as neural networks offer a ubiquitous method to develop models for physical processes. Limiting what kinds of microstructural information is used as input and assessing normalized changes in network error, the relative importance of different mechanisms can be inferred. Here we utilize binned, initial density information as network inputs to predict local shock heating in a granular high explosive trained from large scale, molecular dynamics simulations. The spatial extend of the density field used in the network is altered to assess the importance and relevant length scales of the physical mechanisms in play, where different microstructural features result in different predictive capability. read less
NOT USED (low confidence) H. Wang, J.-H. Lee, J. H. Kim, and H. Shin, “Multiscale strategy to predict the fracture toughness and crack extension behavior of ozone-functionalized carbon nanotube/epoxy nanocomposites,” Chemical Engineering Journal. 2023. link Times cited: 3
NOT USED (low confidence) D. Xu, H. Wan, X. Yao, J. Li, and L.-T. Yan, “Molecular Simulations in Macromolecular Science,” Chinese Journal of Polymer Science. 2023. link Times cited: 3
NOT USED (low confidence) J. Li, P. Heng, B. Wang, B. Wang, N. Liu, and X. Wang, “Comparative Study on the Unimolecular Decompositions of Energetic Regioisomers: BFTF-1 and BFTF-2,” FirePhysChem. 2023. link Times cited: 0
NOT USED (low confidence) B. Hamilton, P. Yoo, M. Sakano, M. M. Islam, and A. Strachan, “High-pressure and temperature neural network reactive force field for energetic materials.,” The Journal of chemical physics. 2023. link Times cited: 2
Abstract: Reactive force fields for molecular dynamics have enabled a … read more
Abstract: Reactive force fields for molecular dynamics have enabled a wide range of studies in numerous material classes. These force fields are computationally inexpensive compared with electronic structure calculations and allow for simulations of millions of atoms. However, the accuracy of traditional force fields is limited by their functional forms, preventing continual refinement and improvement. Therefore, we develop a neural network-based reactive interatomic potential for the prediction of the mechanical, thermal, and chemical responses of energetic materials at extreme conditions. The training set is expanded in an automatic iterative approach and consists of various CHNO materials and their reactions under ambient and shock-loading conditions. This new potential shows improved accuracy over the current state-of-the-art force fields for a wide range of properties such as detonation performance, decomposition product formation, and vibrational spectra under ambient and shock-loading conditions. read less
NOT USED (low confidence) S. Feng et al., “Effect of neutron irradiation on structure and decomposition of α-RDX: A ReaxFF molecular dynamics study,” Computational and Theoretical Chemistry. 2023. link Times cited: 1
NOT USED (low confidence) S. Liu, L. Wei, Q. Zhou, T. Yang, S.-bai Li, and Q. Zhou, “Simulation Strategies for ReaxFF Molecular Dynamics in Coal Pyrolysis Applications: A Review,” Journal of Analytical and Applied Pyrolysis. 2023. link Times cited: 16
NOT USED (low confidence) H. Wei, T. Li, K. Yao, and Z. Xuan, “ReaxFF molecular dynamics simulations on thermal decomposition of RDX-based CMDB propellants,” Journal of Molecular Modeling. 2022. link Times cited: 0
NOT USED (low confidence) J. Jiang, L. Song, F. Zhao, S. Xu, and X. Ju, “Molecular evolution mechanisms of FOX-7 under high-pressure shock on different crystal faces,” Energetic Materials Frontiers. 2022. link Times cited: 0
NOT USED (low confidence) J. Quansah, X.-X. Zhang, Q. Wasiullah, and Q. Yan, “Mechanical and Thermophysical Properties of Energetic Crystals: Evaluation Methods and Recent Achievements,” FirePhysChem. 2022. link Times cited: 6
NOT USED (low confidence) A. Chaurasia, S. Jalan, and A. Parashar, “An atomistic approach to study the dynamic and structural response in 2D nanofiller reinforced polyethylene nanocomposites under ultra-short shock pulse loading,” Mechanics of Materials. 2022. link Times cited: 6
NOT USED (low confidence) K. Tan et al., “Impacts of defect distribution on the ignition of crystalline explosives: An insight from the overlapping effect,” Energetic Materials Frontiers. 2022. link Times cited: 2
NOT USED (low confidence) С. А. Губин, С. А. Козлова, and И. В. Маклашова, “ПОЛУЧЕНИЕ ИЗОТЕРМИЧЕСКИХ ХАРАКТЕРИСТИК, ПАРАМЕТРОВ УРАВНЕНИЯ СОСТОЯНИЯ ДЛЯ PETN МЕТОДАМИ РЕАКЦИОННОЙ МОЛЕКУЛЯРНОЙ ДИНАМИКИ И ТЕМОДИНАМИКИ,” Gorenie i vzryv (Moskva) - Combustion and Explosion. 2022. link Times cited: 0
Abstract: Методом молекулярной динамики (МД) в программном пакете LAMM… read more
Abstract: Методом молекулярной динамики (МД) в программном пакете LAMMPS (Large-scale Atom-ic/Molecular Massively Parallel Simulator) с использованием реакционного силового поля ReaxFF-lg были рассчитаны изотермы нереагирующего вещества PETN в диапазоне давлений до 30 ГПа. Были получены значения коэффициента модуля всестороннего сжатия Ко = 9,6 ГПа и производной модуля сжатия Ко по давлению К'о = 8,0, которые можно использовать как параметры термического уравнения Берча-Мурнагана 3-го порядка. Были подобраны коэффициенты уравнения состояния (УрС) в форме Ми-Грюнайзена, часто используемого для моделирования теплофизических свойств вещества, в том числе при статическом и ударно-волновом сжатии. Для нахождения коэффициентов УрС применялся метод построения изохорно-изотермического потенциала твердых веществ в форме квазигармонического приближения Эйнштейна. Верификация полученных результатов показала хорошее согласие с экспериментальными данными в широком диапазоне изменения давления и температуры, в том числе вдоль ударной адиабаты. read less
NOT USED (low confidence) J. Zhao, Z. Huang, G. Jin, M. Gao, and H. Zhu, “Reactive Molecular Dynamics Calculation and Ignition Delay Test of the Mixture of an Additive and 2-Azido-N,N-dimethylethanamine with Dinitrogen Tetroxide,” ACS Omega. 2022. link Times cited: 0
Abstract: In order to shorten the ignition delay of 2-azido-N,N-dimeth… read more
Abstract: In order to shorten the ignition delay of 2-azido-N,N-dimethylethanamine (DMAZ) and dinitrogen tetroxide (NTO), four amines [tert-butylamine, pyrrole, N,N,N′,N′-tetramethyl ethylenediamine (TMEDA), and diethylenetriamine (DABH)] with a mass fraction of 5% were added to DMAZ, and the potential energy change and the product change during the reaction of the mixture of an additive and DMAZ with NTO were analyzed by Reactive molecular dynamics (ReaxFF MD) calculation. Then, the ignition delay of the mixture of the additive and DMAZ as well as pure DMAZ with NTO was measured by a drop experiment with a photoelectric sensor and high-speed camera. The results show that the addition of pyrrole greatly reduced the time to reach the maximum system energy and greatly increased the rate of HNO2 formation. The dripping of the fuel was approximately a uniform linear motion, and the expression was y = 43.13 + 7.16x. The ignition delay time recorded by the camera was in good agreement with that of the optical signal. The measured ignition delay time for DMAZ with NTO was 261.5 ms. The mixture of pyrrole and DMAZ with NTO had the shortest ignition delay time of 100 ms, and the proportion of shortening the ignition delay time was the largest. The results of the droplet experiment were consistent with those of ReaxFF MD simulation, indicating that HNO2 plays an important role in the ignition delay, that is, the formation rate of HNO2 is positively correlated with the ignition delay. read less
NOT USED (low confidence) W. Qian, X. Xue, J. Liu, and C. Zhang, “Molecular Forcefield Methods for Describing Energetic Molecular Crystals: A Review,” Molecules. 2022. link Times cited: 6
Abstract: Energetic molecular crystals are widely applied for military… read more
Abstract: Energetic molecular crystals are widely applied for military and civilian purposes, and molecular forcefields (FF) are indispensable for treating the microscopic issues therein. This article reviews the three types of molecular FFs that are applied widely for describing energetic crystals—classic FFs, consistent FFs, and reactive FFs (ReaxFF). The basic principle of each type of FF is briefed and compared, with the application introduced, predicting polymorph, morphology, thermodynamics, vibration spectra, thermal property, mechanics, and reactivity. Finally, the advantages and disadvantages of these FFs are summarized, and some directions of future development are suggested. read less
NOT USED (low confidence) B. Hamilton and A. Strachan, “Many-Body Mechanochemistry: Intra-molecular Strain in Condensed Matter Chemistry,” Physical Review Materials. 2022. link Times cited: 8
Abstract: Mechanical forces acting on atoms or molecular groups can al… read more
Abstract: Mechanical forces acting on atoms or molecular groups can alter chemical kinetics and decomposition paths. So called mechanochemistry has been proposed to influence a variety of processes, from the formation of prebiotic compounds during planetary collisions to the shock-induced initiation of explosives. It has also been harnessed in various engineering applications such as mechanophores and ball milling in industrial applications. Experimental and computational tools designed to characterize the effect of mechanics on chemistry have focused exclusively on simple linear forces between pairs of atoms or molecular groups. However, the mechanical loading in condensed matter systems is significantly more complex and involves many-body deformations. Therefore, we propose a methodology to characterize the effect of many-body intra-molecular strains on decomposition kinetics and reaction pathways. We combine four-body external potentials with reactive molecular dynamics and show that many body strains that mimic those observed in condensed matter encourage bond rupture in a spiropyran mechanophore and accelerate thermal decomposition of condensed TATB, an energetic material. The approach is generalizable to a variety of systems and can be used in conjunction with ab initio molecular dynamics, and the two examples utilized here illustrates both the versatility of the method and the importance of many-body mechanochemistry. read less
NOT USED (low confidence) K. Yang, L. Chen, D.-yang Liu, D. Geng, J. Lu, and J. Wu, “Quantitative prediction and ranking of the shock sensitivity of explosives via reactive molecular dynamics simulations,” Defence Technology. 2022. link Times cited: 3
NOT USED (low confidence) S. Yu, R. Chu, X. Li, G. Wu, and X. Meng, “Combined ReaxFF and Ab Initio MD Simulations of Brown Coal Oxidation and Coal–Water Interactions,” Entropy. 2021. link Times cited: 4
Abstract: In this manuscript, we use a combination of Car–Parrinello m… read more
Abstract: In this manuscript, we use a combination of Car–Parrinello molecular dynamics (CPMD) and ReaxFF reactive molecular dynamics (ReaxFF-MD) simulations to study the brown coal–water interactions and coal oxidation. Our Car–Parrinello molecular dynamics simulation results reveal that hydrogen bonds dominate the water adsorption process, and oxygen-containing functional groups such as carboxyl play an important role in the interaction between brown coal and water. The discrepancy in hydrogen bonds formation between our simulation results by ab initio molecular dynamics (CPMD) and that by ReaxFF-MD indicates that the ReaxFF force field is not capable of accurately describing the diffusive behaviors of water on lignite at low temperatures. The oxidations of brown coal for both fuel rich and fuel lean conditions at various temperatures were investigated using ReaxFF-MD simulations through which the generation rates of major products were obtained. In addition, it was observed that the density decrease significantly enhances the generation of gaseous products due to the entropy gain by reducing system density. Although the ReaxFF-MD simulation of complete coal combustion process is limited to high temperatures, the combined CPMD and ReaxFF-MD simulations allow us to examine the correlation between water adsorption on brown coal and the initial stage of coal oxidation. read less
NOT USED (low confidence) Y. Li et al., “Molecular Dynamics Simulations of the Thermal Decomposition of 3,4-Bis(3-nitrofurazan-4-yl)furoxan,” ACS Omega. 2021. link Times cited: 2
Abstract: When stimulated, for example, by a high temperature, the phy… read more
Abstract: When stimulated, for example, by a high temperature, the physical and chemical properties of energetic materials (EMs) may change, and, in turn, their overall performance is affected. Therefore, thermal stability is crucial for EMs, especially the thermal dynamic behavior. In the past decade, significant efforts have been made to study the thermal dynamic behavior of 3,4-bis(3-nitrofurazan-4-yl)furoxan (DNTF), one of the new high-energy-density materials (HEDMs). However, the thermal decomposition mechanism of DNTF is still not specific or comprehensive. In this study, the self-consistent-charge density-functional tight-binding method was combined with molecular dynamics (MD) simulations to reveal the differences in the thermal decomposition of DNTF under four heating conditions. The O–N (O) bond would fracture first during DNTF initial thermal decomposition at medium and low temperatures, thus triggering the cracking of the whole structure. At 2000 and 2500 K, NO2 loss on outer ring I is the fastest initial thermal decomposition pathway, and it determines that the decomposition mechanism is different from that of a medium-low temperature. NO2 is found to be the most active intermediate product; large molecular fragments, such as C2N2O, are found for the first time. Hopefully, these results could provide some insights into the decomposition mechanism of new HEDMs. read less
NOT USED (low confidence) J. Wang, B. Zhu, and Y. Sun, “Microscopic mechanism of α-rhombic crystal boron nanocluster oxidation in oxygen,” Fuel. 2021. link Times cited: 10
NOT USED (low confidence) L. Yi et al., “Molecular dynamic study on hydrogen production from unsymmetrical dimethylhydrazine in supercritical water,” Journal of Molecular Liquids. 2021. link Times cited: 7
NOT USED (low confidence) G. Fiorin, M. J. DelloStritto, S. Percec, and M. Klein, “Shear response in crystalline models of poly(p-phenylene terephthalamide),” Molecular Physics. 2021. link Times cited: 3
Abstract: ABSTRACT The high anisotropy of polymer-based fibres confers… read more
Abstract: ABSTRACT The high anisotropy of polymer-based fibres confers them high tensile strength, but also makes them more vulnerable against non-uniform mechanical loads. This is even more important for Kevlar® fibres, which are made up of individual fibrils containing crystalline domains at different orientations. In this work, crystals of poly(p-phenylene terephthalamide), or PPTA, are subject to shear strain and their response simulated in atomic detail. For shear deformations involving movements orthogonal to the chains’ axis, an originally defect-free crystal fully recovers its native contacts and original shear strength after repeated failures. Full recovery of crystalline contacts proceeds over tens of nanoseconds, demonstrating the importance of sampling realistic strain rates. For shear deformations involving movements parallel to the chains’ axis, the crystal accumulates an increasing number of defects that lower its shear strength. Although the same types of intermolecular forces make up the response of a PPTA crystal to each shear mode, the relative contributions of these modes in a specific type of applied load will affect profoundly how Kevlar® fibrils and fibres fail under shear. The shear stress–strain profiles here computed will ultimately benefit the development of quantitative mechanical models of Kevlar® as well as new polyamide materials. GRAPHICAL ABSTRACT read less
NOT USED (low confidence) L. Jiang et al., “Study on N-guanylurea-dinitramide (GUDN) decomposition using theoretical simulations, online photoionization mass spectrometry and TG-DSC-IR-MS experiments,” Combustion and Flame. 2021. link Times cited: 9
NOT USED (low confidence) G. Lan et al., “Thermal decomposition mechanism study of 3-nitro-1,2,4-triazol-5-one (NTO): Combined TG-FTIR-MS techniques and ReaxFF reactive molecular dynamics simulations,” Fuel. 2021. link Times cited: 39
NOT USED (low confidence) N. Goga et al., “A Review of Recent Developments in Molecular Dynamics Simulations of the Photoelectrochemical Water Splitting Process,” Catalysts. 2021. link Times cited: 8
Abstract: In this review, we provide a short overview of the Molecular… read more
Abstract: In this review, we provide a short overview of the Molecular Dynamics (MD) method and how it can be used to model the water splitting process in photoelectrochemical hydrogen production. We cover classical non-reactive and reactive MD techniques as well as multiscale extensions combining classical MD with quantum chemical and continuum methods. Selected examples of MD investigations of various aqueous semiconductor interfaces with a special focus on TiO2 are discussed. Finally, we identify gaps in the current state-of-the-art where further developments will be needed for better utilization of MD techniques in the field of water splitting. read less
NOT USED (low confidence) L. Shen et al., “A Molecular‐Level Interface Design Enabled High‐Strength and High‐Toughness Carbon Nanotube Buckypaper,” Macromolecular Materials and Engineering. 2021. link Times cited: 2
NOT USED (low confidence) B. Zhu, L. Zhu, Y. Wan, S. Deng, C. Zhang, and J. Luo, “Multicomponent metal-organic frameworks with aggregation-induced emission characteristics as fluorescence sensor array for the identification of energetic compounds,” Sensors and Actuators B-chemical. 2021. link Times cited: 13
NOT USED (low confidence) M. S. Islam, I. Mia, S. Ahammed, C. Stampfl, and J. Park, “Exceptional in-plane and interfacial thermal transport in graphene/2D-SiC van der Waals heterostructures,” Scientific Reports. 2020. link Times cited: 18
NOT USED (low confidence) S. Kozlova, S. Gubin, and I. Maklashova, “Simulation of the isothermal and Hugoniot characteristics of organic compounds via the reactive molecular dynamics,” Journal of Physics: Conference Series. 2020. link Times cited: 0
Abstract: This article provides the results of reactive molecular dyna… read more
Abstract: This article provides the results of reactive molecular dynamics simulation of shock loading of cyclic hydrocarbon C6H6 and isotherm curve of energy-intensive nitramine C4H8N8O8. To describe the interatomic interaction, as well as to analyze the kinetics of decomposition, we used two parameterizations of the reactive force field ReaxFF. In this paper, we compared the capabilities of ReaxFF force fields to describe the kinetics of the decomposition of benzene behind the front of a shock wave and the ability to reproduce phase transformations of carbon under conditions of high pressures and temperatures. The results obtained are in good agreement with the experiment. read less
NOT USED (low confidence) Y.-E. Liu, J. Hu, H. Hou, and B. Wang, “Development and application of a ReaxFF reactive force field for molecular dynamics of perfluorinatedketones thermal decomposition,” Chemical Physics. 2020. link Times cited: 12
NOT USED (low confidence) J. Zeng, L. Zhang, H. Wang, T. Zhu, and T. Zhu, “Explore the Chemical Space of Linear Alkanes Pyrolysis via Deep Potential Generator.” 2020. link Times cited: 13
Abstract:
Reactive molecular dynamics (MD) simulation is a power… read more
Abstract:
Reactive molecular dynamics (MD) simulation is a powerful tool to study the reaction mechanism of complex chemical systems. Central to the method is the potential energy surface (PES) that can describe the breaking and formation of chemical bonds. The development of PES of both accurate and efficent has attracted significant effort in the past two decades. Recently developed Deep Potential (DP) model has the promise to bring ab initio accuracy to large-scale reactive MD simulations. However, for complex chemical reaction processes like pyrolysis, it remains challenging to generate reliable DP models with an optimal training dataset. In this work, a dataset construction scheme for such a purpose was established. The employment of a concurrent learning algorithm allows us to maximize the exploration of the chemical space while minimize the redundancy of the dataset. This greatly reduces the cost of computational resources required by ab initio calculations. Based on this method, we constructed a dataset for the pyrolysis of n-dodecane, which contains 35,496 structures. The reactive MD simulation with the DP model trained based on this dataset revealed the pyrolysis mechanism of n-dodecane in detail, and the simulation results are in good agreement with the experimental measurements. In addition, this dataset shows excellent transferability to different long-chain alkanes. These results demonstrate the advantages of the proposed method for constructing training datasets for similar systems.
read less
NOT USED (low confidence) B. Rice, W. Mattson, J. Larentzos, and E. Byrd, “Heuristics for chemical species identification in dense systems.,” The Journal of chemical physics. 2020. link Times cited: 6
Abstract: A new approach to identify chemical species from molecular d… read more
Abstract: A new approach to identify chemical species from molecular dynamics (MD) simulations of reacting materials under extreme temperatures and pressures is presented. The approach is based on bond-distance and vibrational criteria, derived from the examination of atomic behavior during a density functional theory MD simulation of an overdriven shock of the explosive pentaerythritol tetranitrate. For comparison, the trajectory was analyzed using popular bonding criteria commonly used in analysis of reactive MD simulations, including distance, distance-time, and bond-order criteria. Cluster analyses using the new time-dependent bond definition approach presented here and a bond-order approach revealed that species and their corresponding lifetimes were strongly dependent on the chosen approach, indicating significant implications for the development of chemical mechanisms and chemical kinetics models using the results of reactive MD simulations. read less
NOT USED (low confidence) Y.-E. Liu, J. Hu, H. Hou, and B. Wang, “ReaxFF reactive force field development and application for molecular dynamics simulations of heptafluoroisobutyronitrile thermal decomposition,” Chemical Physics Letters. 2020. link Times cited: 4
NOT USED (low confidence) S.-jie Zhang, Z. Gao, Q. Jia, N. Liu, J. Zhang, and K. Kou, “Fabrication and characterization of surface modified HMX@PANI core-shell composites with enhanced thermal properties and desensitization via in situ polymerization,” Applied Surface Science. 2020. link Times cited: 35
NOT USED (low confidence) N. Dasgupta, Y. Shin, M. Fedkin, and A. V. van Duin, “ReaxFF molecular dynamics simulations of electrolyte-water systems at supercritical temperature.,” The Journal of chemical physics. 2020. link Times cited: 8
Abstract: We have performed ReaxFF molecular dynamics simulations of a… read more
Abstract: We have performed ReaxFF molecular dynamics simulations of alkali metal-chlorine pairs in different water densities at supercritical temperature (700 K) to elucidate the structural and dynamical properties of the system. The radial distribution function and the angular distribution function explain the inter-ionic structural and orientational arrangements of atoms during the simulation. The coordination number of water molecules in the solvation shell of ions increases with an increase in the radius of ions. We find that the self-diffusion coefficient of metal ions increases with a decrease in density under supercritical conditions due to the formation of voids within the system. The hydrogen bond dynamics has been interpreted by the residence time distribution of various ions, which shows Li+ having the highest water retaining capability. The void distribution within the system has been analyzed by using the Voronoi polyhedra algorithm providing an estimation of void formation within the system at high temperatures. We observe the formation of salt clusters of Na+ and K+ at low densities due to the loss of dielectric constants of ions. The diffusion of ions gets altered dramatically due to the formation of voids and nucleation of ions in the system. read less
NOT USED (low confidence) D.-yang Liu et al., “Decomposition and Energy-Enhancement Mechanism of the Energetic Binder Glycidyl Azide Polymer at Explosive Detonation Temperatures.,” The journal of physical chemistry. A. 2020. link Times cited: 11
Abstract: Replacing existing inert binders with energetic ones in comp… read more
Abstract: Replacing existing inert binders with energetic ones in composite explosives is a novel way to improve the explosive performance, on the proviso that energetic binders are capable of releasing chemical energy rapidly in the detonation environment. Known to be a promising candidate, the reaction mechanism of glycidyl azide polymer (GAP) at typical detonation temperatures higher than 3000K has been theoretically studied in this work at the atomistic level. By analyzing and tracking the cleavage of characteristic chemical bonds, it was found that at the detonation temperature, GAP was able to release a large amount of energy and small molecule products at a comparable speed to commonly used explosives in the early reaction stage, which was mainly attributed to the decomposition of azide groups into N2 and the main chain breakage into small fragments. Moreover, N2 generation was found to be accelerated by H atom transfer at an earlier reaction step. The dissociation energy of the main chain was lowered with structure deformation so as to facilitate the fragmentation of the GAP chain. Based on this analytical study of reaction kinetics, GAP was found to have higher reactivity at the detonation temperature than at lower temperatures. The small molecules yield rate is as the same order of magnitude as an explosive detonation reaction, indicating that GAP has the potential to improve the performance of composite explosives. Our study reveals the chemical decomposition mechanism of a typical energetic binder, which would aid in the future design and synthesis of energetic binders so as to achieve both sensitivity-reducing and energy-enhancing performance goals simultaneously. read less
NOT USED (low confidence) Y.-E. Liu, H. Hou, and B. Wang, “Thermal decomposition of vegetable insulating oils from reactive molecular dynamics,” Chemical Physics Letters. 2020. link Times cited: 2
NOT USED (low confidence) A. Saha and A. Das, “Dynamical behavior of nonlinear wave solutions of the generalized Newell–Whitehead–Segel equation,” International Journal of Modern Physics C. 2020. link Times cited: 5
Abstract: Dynamical behavior of nonlinear wave solutions of the pertur… read more
Abstract: Dynamical behavior of nonlinear wave solutions of the perturbed and unperturbed generalized Newell–Whitehead–Segel (GNWS) equation is studied via analytical and computational approaches for the fir... read less
NOT USED (low confidence) J. Yuan, H. Ren, Y. Wei, W.-S. Xu, G. Ji, and D. Wei, “The Reaction and Microscopic Electron Properties from Dynamic Evolutions of Condensed-Phase RDX Under Shock Loading,” Zeitschrift für Naturforschung A. 2020. link Times cited: 0
Abstract: Microscopic electron properties of α-hexahydro-1,3,5-trinitr… read more
Abstract: Microscopic electron properties of α-hexahydro-1,3,5-trinitro-1,3,5-triazine (α-RDX) with different shock wave velocities have been investigated based on molecular dynamics together with multi-scale shock technique. The studied shock wave velocities are 8, 9 and 10 km ⋅ s−1. It has been said that the shock sensitivity and reaction initiation of explosives are closely relevant with their microscopic electron properties. The reactions, including the reaction products, which are counted from the trajectory during the simulations are analysed first. The results showed that the number of the products strictly rely on shock wave velocities. The reaction rates and decomposition rates are also studied, which showed the differences between the different shock velocities. The results of electron properties show that α-RDX is a wide-gap insulator in the ground state and the metallisation conditions of shocked RDX are determined, which are lower than under-static high pressure. read less
NOT USED (low confidence) N. Dasgupta, Y. K. Shin, M. Fedkin, and A. V. van Duin, “ReaxFF molecular dynamics simulations on the structure and dynamics of electrolyte water systems at ambient temperature,” Computational Materials Science. 2020. link Times cited: 17
NOT USED (low confidence) D. Hu, X. Gu, B. Cui, J. Pei, and Q. Zhang, “Modeling the Oxidative Aging Kinetics and Pathways of Asphalt: A ReaxFF Molecular Dynamics Study,” Energy & Fuels. 2020. link Times cited: 56
Abstract: The ReaxFF molecular dynamics simulations, which can predict… read more
Abstract: The ReaxFF molecular dynamics simulations, which can predict chemical reactions, were performed on integral asphalt and individual asphalt molecules at different temperatures and oxygen levels to i... read less
NOT USED (low confidence) A. Islam, M. S. Islam, N. Ferdous, J. Park, A. G. Bhuiyan, and A. Hashimoto, “Anisotropic mechanical behavior of two dimensional silicon carbide: effect of temperature and vacancy defects,” Materials Research Express. 2019. link Times cited: 27
Abstract: Mechanical stability, which is featured by high tensile stre… read more
Abstract: Mechanical stability, which is featured by high tensile strength, is one of the most critical concerns for the reliability of next-generation nanoelectromechanical systems (NEMS). Presently, sp2 hybridized two-dimensional silicon carbide (2D-SiC) is supposed to be a novel nanomaterial to apply in nanocomposites, NEMS, and nano-energy harvesting applications because of its amazing electronic, mechanical and thermal properties. This paper explores the mechanical behavior, including fracture stress, fracture strain, and elastic modulus of both pristine and vacancy defected 2D-SiC at temperatures 300–700 K using molecular dynamics simulation. Two types of vacancy defects such as point and bi-vacancies with concentration 0.1%–1.0% are considered. Moreover, the effect of system size and strain rate on the mechanical behavior of 2D-SiC is also analyzed. A highly anisotropic mechanical behavior is found at all temperature and defect conditions. At 300 K, a fracture stress and an elastic modulus of 71.02 GPa and 637.26 GPa, respectively is obtained along the armchair direction, which is ∼24.42% and ∼14.38% higher compared to the zigzag directed fracture stress and elastic modulus. A reduction of fracture stress, fracture strain, and elastic modulus with the increase of temperature and defect concentration is also perceived in both armchair and zigzag directions. Moreover, due to the large symmetry breakdown by the point vacancy, a comparatively larger drastic reduction is noticed in the fracture behavior than the bi-vacancy at all temperatures and loading directions. These results would provide a new insight for solving the mechanical instability problem of SiC-based NEMS and nanodevices in the near future. read less
NOT USED (low confidence) D. Furman and D. Wales, “Transforming the Accuracy and Numerical Stability of ReaxFF Reactive Force Fields.,” The journal of physical chemistry letters. 2019. link Times cited: 16
Abstract: Molecular dynamics (MD) simulations provide an important lin… read more
Abstract: Molecular dynamics (MD) simulations provide an important link between theories and experiments. While ab initio methods can be prohibitively costly, the ReaxFF force field has facilitated in silico studies of chemical reactivity in complex, condensed-phase systems. However, the relatively poor energy conservation in ReaxFF MD has either limited the applicability to short time scales, in cases where energy propagation is important, or has required a continuous coupling of the system to a heat bath. In this study, we reveal the root cause of the unsatisfactory energy conservation, and offer a straightforward solution. The new scheme results in orders of magnitude improvement in energy conservation, numerical stability, and accuracy of ReaxFF force fields, compared to the previous state-of-the-art, at no additional cost. We anticipate that these improvements will open new avenues of research for more accurate reactive simulations in complex systems on long time scales. read less
NOT USED (low confidence) Z. Zheng, H. Zhan, Y. Nie, X. Xu, and Y. T. Gu, “Role of Nitrogen on the Mechanical Properties of the Novel Carbon Nitride Nanothreads,” The Journal of Physical Chemistry C. 2019. link Times cited: 7
Abstract: Carbon nanothread (C-NTH) is a new ultrathin one-dimensional… read more
Abstract: Carbon nanothread (C-NTH) is a new ultrathin one-dimensional sp3 carbon nanostructure, which exhibits promising applications in novel carbon nanofibers and nanocomposites. Recently, researchers have successfully developed a new alternative structure - ultrathin carbon nitride nanothread (CN-NTH). In this work, we investigate the mechanical properties of CN-NTHs through large-scale molecular dynamics simulations. Comparing with their C-NTH counterparts, CN-NTHs are found to exhibit a higher tensile and bending stiffness. In particular, because of the bond redistribution, the CN-NTHs in the polymer I group and tube (3,0) group are found to possess a higher failure strain than their C-NTH counterparts. However, the CN-NTH in the polytwistane group has a smaller failure strain compared with the pristine C-NTH. According to the atomic configurations, the presence of nitrogen atoms always leads to stress/strain concentrations for the nanothreads under tensile deformation. This study provides a comprehensive understanding of the mechanical properties of CN-NTHs, which should shed light on their potential applications such as fibers or reinforcements for nanocomposites. read less
NOT USED (low confidence) J. Wang, G.-J. Guo, Y. Han, Q. Hou, M. Geng, and Z. Zhang, “Mechanolysis mechanisms of the fused aromatic rings of anthracite coal under shear stress,” Fuel. 2019. link Times cited: 29
NOT USED (low confidence) D. Hwang et al., “3D graphene-cellulose nanofiber hybrid scaffolds for cortical reconstruction in brain injuries,” 2D Materials. 2019. link Times cited: 12
Abstract: Designing the nature-driven 3D scaffold is essential for rec… read more
Abstract: Designing the nature-driven 3D scaffold is essential for reconstructing of the injured brain in association with stem cell replacement therapy. In this paper, we developed brain cortex-mimetic 3D hybrid scaffolds and applied them to a motor-cortectomy rat model. Graphene oxide bacterial cellulose (GO-BC) hybrid scaffold integrated GOs stably and homogeneously within BC nanofibrous building blocks made of BC and amphiphilic comb-like polymers (APCLP). Density functional theory calculations and molecular dynamics simulations revealed higher binding energies between GO-BC and APCLP than between GO or APLCP with BC. The monodispersed human neural stem cells (F3 cells) incorporated within the GO-BC scaffold generated a large number of differentiated neurons with robust neurite outgrowths and possible synapse formation in vitro. In corticectomized rats and nude mice, highly sensitive photoacoustic signals visualized the GO-BC at the implant site. Moreover, the implanted F3 cells within GO-BC were found to survive/proliferate and differentiate to neuronal lineage from the showing neuronal and synapse markers shown on ex vivo immunofluorescence staining in bioluminescence imaging. Cortex-mimetic and stem cell-instructive monodisperse GO-BC hybrid scaffolds are likely to be appropriate nanoplatforms for stem cell implantation to reconstruct injured/lost brain tissues and actively differentiate neural stem cells. read less
NOT USED (low confidence) D. Depew et al., “Thermal Decomposition of Hydroxylammonium Nitrate: ReaxFF Training Set Development for Molecular Dynamics Simulations,” AIAA Propulsion and Energy 2019 Forum. 2019. link Times cited: 1
NOT USED (low confidence) Y. Ma et al., “Enhanced thermal resistance of carbon/phenolic composites by addition of novel nano-g-C3N4,” Composites Science and Technology. 2019. link Times cited: 21
NOT USED (low confidence) L. Dresselhaus-Cooper et al., “Pressure-Thresholded Response in Cylindrically Shocked Cyclotrimethylene Trinitramine (RDX).,” The journal of physical chemistry. A. 2019. link Times cited: 5
Abstract: We demonstrate a strongly thresholded response in cyclotrime… read more
Abstract: We demonstrate a strongly thresholded response in cyclotrimethylene trinitramine (RDX) when it is cylindrically shocked using a novel waveguide geometry. Using ultrafast single-shot multi-frame imaging, we demonstrate that <100-μm diameter single crystals of RDX embedded in a polymer host deform along preferential planes for >100 ns after the shock first arrives in the crystal. We use in-situ imaging and time-resolved photoemission to demonstrate that short-lived chemistry is linked to high-energy deformation planes. Using scanning electron microscopy and ultrasmall-angle X-ray scattering, we demonstrate that the shock-induced dynamics leave behind sintered crystals, with pore shapes and sizes that change significantly with shock energy. A threshold pressure of ~ 12 GPa at the center of convergence separated the single-mode planar crystal deformations from the chemistry-coupled multi-plane dynamics at higher pressures. Our observations indicate preferential deformation mechanics in our cylindrically shocked system, despite the applied stress along many different crystallographic planes. read less
NOT USED (low confidence) Y.-L. Liu, X. Zhang, and J. Ding, “Chemical effect of NO on CH4 oxidation during combustion in O2/NO environments,” Chemical Physics Letters. 2019. link Times cited: 8
NOT USED (low confidence) M. Chen, Y. Zhu, J. Xia, and H. Wu, “Molecular insights into the initial formation of pyrolytic carbon upon carbon fiber surface,” Carbon. 2019. link Times cited: 24
NOT USED (low confidence) L. Xie, Y. Shao, W. Zhong, H. Ben, and K.-xi Li, “Molecular dynamic simulation on the oxidation process of coal tar pitch,” Fuel. 2019. link Times cited: 23
NOT USED (low confidence) D.-yang Liu, L. Chen, D. Geng, J. Lu, and J. Wu, “Correlation between Chemical Bond Cleavage and Detonation of ε-2,4,6,8,10,12-Hexanitrohexaazaisowurtzitane,” The Journal of Physical Chemistry C. 2019. link Times cited: 15
Abstract: Researchers have been striving to determine the connection b… read more
Abstract: Researchers have been striving to determine the connection between the microscopic chemical reactions and macroscopic detonation laws of explosives. In this study, we performed reactive molecular dynamics simulations of the shock-induced explosion of the 2,4,6,8,10,12-hexanitrohexaazaisowurtzitane explosive. The results show that detonation is mainly determined by the rapid irreversible cleavage of the C–N and C–H bonds. Such C–N and C–H bond cleavages determine the early formation of N2 and H2O. The detonation reaction occurs when the cleavage rates exceed 3.11 and 4.15%/ps for the C–N and C–H bonds, respectively. A higher shock velocity results in higher cleavage rates of these bonds, but it also leads to more atoms being trapped in clusters. However, the decomposition rate of these clusters is mainly affected by the decrease in the density, not by the shock velocity, indicating that the late detonation reaction is mainly based on the characteristics of the explosive. read less
NOT USED (low confidence) L. Mejía and I. Franco, “Force–conductance spectroscopy of a single-molecule reaction,” Chemical Science. 2019. link Times cited: 7
Abstract: We demonstrate how simultaneous measurements of conductance … read more
Abstract: We demonstrate how simultaneous measurements of conductance and force can be used to monitor the step-by-step progress of a mechanically activated cis-to-trans isomerization single-molecule reaction, including events that cannot be distinguished using force or conductance alone. read less
NOT USED (low confidence) L. Linlin and S. Hu, “Ab initio calculations of the NO2 fission for CL-20 conformers,” Journal of Energetic Materials. 2018. link Times cited: 5
Abstract: ABSTRACT NO2 fission is regarded to be the most important in… read more
Abstract: ABSTRACT NO2 fission is regarded to be the most important initial decomposition process of 2,4,6,8,10,12-hexanitro-2,4,6,8,10,12-hexaazaisowurtzitane (CL-20). In this study, four CL-20 conformers based on the ε-CL-20 were obtained after the optimization at m062x/cc-pvtz level, and the bond length, bond order and bond dissociation energy of the N-N bonds were examined to investigate the stability of these bonds. In addition, the rate constants and activation energy of the NO2 fission were evaluated using the microcanonical variational transition state theory (μVT). The calculation results have shown that N-N bonds in the case of pseudo-equatorial and axial of nitro groups are the most stable and the least stable, respectively, by evaluating the bond length, bond order and minimum energy path (MEP). The NO2 fission rate constants are affected by not only the stability of N-N bonds but also the repulsion forces from the other nitro groups, and the fission process for pseudo-equatorial positioning of nitro groups is easier to be accelerated due to the increase of the repulsion forces. The decomposition of CL-20 conformer may mainly originate from the fission of the pseudo-equatorial positioning of nitro groups, especially for CL-20 III conformer because of the significant low activation energy. read less
NOT USED (low confidence) M. Sakano, B. Hamilton, M. M. Islam, and A. Strachan, “Role of Molecular Disorder on the Reactivity of RDX,” The Journal of Physical Chemistry C. 2018. link Times cited: 25
Abstract: Shock initiation of heterogeneous high-energy materials is o… read more
Abstract: Shock initiation of heterogeneous high-energy materials is often preceded by the loss of crystalline order around hotspots where mechanical energy is localized and chemical reactions start. We use ... read less
NOT USED (low confidence) D. Xiang and W. Zhu, “Adiabatic and constant volume decomposition process of condensed phase δ-1,3,5,7-tetranitro-1,3,5,7-tetrazocane at high temperatures: Quantum molecular dynamics simulations.,” Journal of molecular graphics & modelling. 2018. link Times cited: 2
NOT USED (low confidence) Z.-Q. Bai, B. Dai, J. Chang, J. Wang, and N. Ge, “Theoretical study for anisotropic responses of the condensed-phase RDX under shock loadings.,” Journal of molecular graphics & modelling. 2018. link Times cited: 3
NOT USED (low confidence) Y. Ma et al., “Extraordinary improvement of ablation resistance of carbon/phenolic composites reinforced with low loading of graphene oxide,” Composites Science and Technology. 2018. link Times cited: 32
NOT USED (low confidence) J. Harrison, J. Schall, S. Maskey, P. Mikulski, M. T. Knippenberg, and B. Morrow, “Review of force fields and intermolecular potentials used in atomistic computational materials research,” Applied Physics Reviews. 2018. link Times cited: 99
Abstract: Molecular simulation is a powerful computational tool for a … read more
Abstract: Molecular simulation is a powerful computational tool for a broad range of applications including the examination of materials properties and accelerating drug discovery. At the heart of molecular simulation is the analytic potential energy function. These functions span the range of complexity from very simple functions used to model generic phenomena to complex functions designed to model chemical reactions. The complexity of the mathematical function impacts the computational speed and is typically linked to the accuracy of the results obtained from simulations that utilize the function. One approach to improving accuracy is to simply add more parameters and additional complexity to the analytic function. This approach is typically used in non-reactive force fields where the functional form is not derived from quantum mechanical principles. The form of other types of potentials, such as the bond-order potentials, is based on quantum mechanics and has led to varying levels of accuracy and transferability. When selecting a potential energy function for use in molecular simulations, the accuracy, transferability, and computational speed must all be considered. In this focused review, some of the more commonly used potential energy functions for molecular simulations are reviewed with an eye toward presenting their general forms, strengths, and weaknesses.Molecular simulation is a powerful computational tool for a broad range of applications including the examination of materials properties and accelerating drug discovery. At the heart of molecular simulation is the analytic potential energy function. These functions span the range of complexity from very simple functions used to model generic phenomena to complex functions designed to model chemical reactions. The complexity of the mathematical function impacts the computational speed and is typically linked to the accuracy of the results obtained from simulations that utilize the function. One approach to improving accuracy is to simply add more parameters and additional complexity to the analytic function. This approach is typically used in non-reactive force fields where the functional form is not derived from quantum mechanical principles. The form of other types of potentials, such as the bond-order potentials, is based on quantum mechanics and has led to varying levels of accuracy and transferabilit... read less
NOT USED (low confidence) K. K. Bejagam, S. Singh, and S. A. Deshmukh, “Nanoparticle activated and directed assembly of graphene into a nanoscroll,” Carbon. 2018. link Times cited: 24
NOT USED (low confidence) P. Krstic, J. Allain, F. J. Domínguez-Gutiérrez, and F. Bedoya, “Unraveling the surface chemistry processes in lithiated and boronized plasma material interfaces under extreme conditions,” Matter and Radiation at Extremes. 2018. link Times cited: 17
NOT USED (low confidence) H. Jiang, Q. Jiao, and C. Zhang, “Early Events When Heating 1,1-Diamino-2,2-dinitroethylene: Self-Consistent Charge Density-Functional Tight-Binding Molecular Dynamics Simulations,” The Journal of Physical Chemistry C. 2018. link Times cited: 26
Abstract: A self-consistent charge density-functional tight-binding me… read more
Abstract: A self-consistent charge density-functional tight-binding method combined with molecular dynamics (MD) simulations and static density functional theory (DFT) calculations is employed to reveal the initial steps responsible for the thermal decay of the low sensitive and high energetic material (EM) 1,1-diamino-2,2-dinitroethylene (FOX-7). Constant temperature heating and temperature-programmed heating are accounted in our MD simulations to resemble the practice. We find that the heating style has an impact on the initial decomposition mechanism of FOX-7. The N–O bond rupture to produce an O radical is observed for the first time in the temperature-programmed heating simulation from 300 to 3000 K, together with the C–NO2 bond cleavage. We also first capture the intra- and intermolecular hydrogen transferred structures in our constant temperature heating simulation at 3000 K, and in this case, the NO2 partition serves as another kind of primary step for thermally decaying FOX-7 too. Static DFT calculations s... read less
NOT USED (low confidence) N. Wang et al., “Dynamic evolution of aluminum nanoparticle impacted by RDX slab,” Chemical Physics Letters. 2018. link Times cited: 4
NOT USED (low confidence) Y. Ma, X. He, L. Meng, X. Xue, and C. Zhang, “Ionization and separation as a strategy for significantly enhancing the thermal stability of an instable system: a case for hydroxylamine-based salts relative to that for pure hydroxylamine.,” Physical chemistry chemical physics : PCCP. 2017. link Times cited: 5
Abstract: Energetic ionic salts (EISs) are attracting extensive attent… read more
Abstract: Energetic ionic salts (EISs) are attracting extensive attention because of their ready preparation and some excellent properties and performances that are comparable to those of common explosives with neutral molecules. Hydroxylamine (HA) is protonated or ionized as H-HA+ and preferred to be introduced into EISs to form HA-based EISs with almost all kinds of anions since these EISs possess higher packing densities and thus more excellent detonation performances than others with the same anions. Moreover, relative to that of pure HA, the thermal stability of HA-based EISs is significantly enhanced. This significantly enhanced thermal stability can extend the application of HA via deprotonation of H-HA+ back to HA; however, the mechanism for stabilization of HA by salification remains unclear. Herein, we employed thermodynamic and kinetic calculations and molecular dynamics simulations to reveal the thermal stability mechanisms of many currently synthesized HA-based EISs and some previously reported EISs with inorganic anions as well as those of pure HA and its aqueous solution. As a result, we have found that the enhanced stability of HA-based EISs is mainly due to the ionization and separation of HA molecules themselves. That is, H-HA+, as an ionized product, is more molecularly stable than HA, with significantly strengthened covalent bonds. The separation of H-HA+ ions or HA molecules makes decomposition more difficult as decomposition initiation varies from bimolecular to unimolecular reactions of HA, with a significant increase in the energy barrier. We have, therefore, proposed a strategy for the stabilization of unstable systems, such as neutral N-rich energetic compounds, by ionization and separation to strengthen these systems and change the decomposition mechanism by increasing the energy barriers of trigger steps such that these barriers become more difficult to overcome, respectively. read less
NOT USED (low confidence) K. Hagita, T. Murashima, H. Takano, and T. Kawakatsu, “Thinning Approximation for Two-Dimensional Scattering Patterns from Coarse-Grained Polymer Melts under Shear Flow,” Journal of the Physical Society of Japan. 2017. link Times cited: 8
Abstract: We proposed a thinning approximation (TA) for estimation of … read more
Abstract: We proposed a thinning approximation (TA) for estimation of the two-dimensional (2D) wide-angle scattering patterns from Kremer–Grest polymer melts under shear. In the TA, extra particles are inserted at the middle of bonds for fine-graining of the coarse-grained polymers. For the case without the TA, spots corresponding to the orientation of bonds at a high shear rate are difficult to observe because the bond length of successive particles is comparable to the distance between neighboring particles. With the insertion of the extra particles, a ring pattern originating from the neighboring particles can be moved to a wide-angle region. Thus, we can observe the spots at high shear rates. We also examined the relationship between 2D scattering patterns and the Weissenberg number, which is defined as the product of the shear rate and the longest relaxation time. It is confirmed that the relationship for coarse-grained polymers with the TA is consistent with that of the all-atomistic model of polyethylene. read less
NOT USED (low confidence) J. A. Martinez, T. Liang, S. Sinnott, and S. Phillpot, “A third-generation charge optimized many body (COMB3) potential for nitrogen-containing organic molecules,” Computational Materials Science. 2017. link Times cited: 11
NOT USED (low confidence) K. L. Joshi and S. Chaudhuri, “Observation of deflagration wave in energetic materials using reactive molecular dynamics,” Combustion and Flame. 2017. link Times cited: 17
NOT USED (low confidence) C. Diao, Y. Dong, and J. Lin, “Reactive force field simulation on thermal conductivities of carbon nanotubes and graphene,” International Journal of Heat and Mass Transfer. 2017. link Times cited: 35
NOT USED (low confidence) M.-Q. Le, “Mechanical properties of penta-graphene, hydrogenated penta-graphene, and penta-CN2 sheets,” Computational Materials Science. 2017. link Times cited: 34
NOT USED (low confidence) W. Zhang and A. V. van Duin, “Second-Generation ReaxFF Water Force Field: Improvements in the Description of Water Density and OH-Anion Diffusion.,” The journal of physical chemistry. B. 2017. link Times cited: 72
Abstract: Hydronium (H3O+) and hydroxide (OH-) ions have anomalously l… read more
Abstract: Hydronium (H3O+) and hydroxide (OH-) ions have anomalously large diffusion constants in aqueous solutions due to their combination of vehicular and Grotthuss hopping diffusion mechanisms. An improvement of the ReaxFF reactive water force field on the basis of our first-generation water force field (water-2010) is presented to describe the proton transfer (PT) mechanisms of H3O+ and OH- in water. Molecular dynamics simulation studies with the water-2017 force field support the Eigen-Zundel-Eigen mechanism for PT in acidic aqueous solution and reproduce the hypercoordinated solvation structure of the OH- in a basic environment. In particular, it predicts the correct order of the diffusion constants of H2O, H3O+, and OH- and their values are in agreement with the experimental data. Another interesting observation is that the diffusion constants of H3O+ and OH- are close to each other at high concentration due to the strong correlation between OH- ions in basic aqueous solution. On the basis of our results, it is shown that ReaxFF provides a novel approach to study the complex acid-base chemical reactions in aqueous solution with any pH value. read less
NOT USED (low confidence) M. Zhong et al., “Influences of different surfaces on anisotropic impact sensitivity of hexahydro-1,3,5-trinitro-1,3,5-triazine,” Vacuum. 2017. link Times cited: 4
NOT USED (low confidence) J. Xu, Y. Bian, Y. Liu, and D. Zhai, “Reactive molecular dynamics study of thermal decomposition of nanocarbon energetic composite materials,” Computational Materials Science. 2017. link Times cited: 9
NOT USED (low confidence) O. Sergeev and A. Yanilkin, “Hydrogen Transfer in Energetic Materials from ReaxFF and DFT Calculations.,” The journal of physical chemistry. A. 2017. link Times cited: 16
Abstract: Energetic materials are characterized by fast and complex ch… read more
Abstract: Energetic materials are characterized by fast and complex chemical reactions. It makes them hardly available for kinetic experiments in relevant conditions and a good target for reactive molecular dynamics simulations. In this work, unimolecular and condensed-phase thermal decomposition of pentaerythritol tetranitrate (PETN) are investigated by ReaxFF molecular dynamics. It is shown that the decomposition kinetics in condensed phase may be described with the activation barrier lower by a factor of 2 than that for isolated molecules. The effect of the intermolecular hydrogen transfer is revealed in condensed phase. Energetic barriers for hydrogen transfer in two energetic materials (methyl nitrate, which is a nitroester as well as PETN, and o-nitrotoluene) are studied with ReaxFF and DFT using nudged elastic band technique. The results indicate that ReaxFF gives significantly lower activation energy for intermolecular hydrogen transfer in nitroesters than different DFT approximations, which explains the molecular dynamics results for PETN. read less
NOT USED (low confidence) M. Wood, M. Cherukara, E. Antillon, and A. Strachan, “Molecular Dynamics Simulations of Shock Loading of Materials: A Review and Tutorial.” 2017. link Times cited: 14
NOT USED (low confidence) Y. Ren, A. Banishev, K. Suslick, J. S. Moore, and D. Dlott, “Ultrafast Proton Transfer in Polymer Blends Triggered by Shock Waves.,” Journal of the American Chemical Society. 2017. link Times cited: 7
Abstract: We describe ultrafast proton transfer in the ground electron… read more
Abstract: We describe ultrafast proton transfer in the ground electronic state triggered by the use of shock waves created by high-speed impacts. The emission of Nile Red (NR), a polarity sensing dye, was used to probe the effects of shock compression in a series of polymers, including polymer Brønsted bases blended with organic acid proton donors. NR undergoes a shock-induced red-shift due to an increase both in density and in polymer polarity. In blends with poly(4-vinylpyridine) (PVP) and phenol, NR showed an excess shock-induced red-shift with a distinct time dependence not present in controls that are incapable of proton transfer. The excess red-shift first appeared with 0.8 km·s-1 impacts. Occurring in ca. 10 ns, this NR red-shift was caused by the formation of an ion pair created by shock-triggered proton transfer from phenol to PVP. read less
NOT USED (low confidence) J. Larentzos and B. Rice, “Transferable Reactive Force Fields: Extensions of ReaxFF-lg to Nitromethane.,” The journal of physical chemistry. A. 2017. link Times cited: 15
Abstract: Transferable ReaxFF-lg models of nitromethane that predict a… read more
Abstract: Transferable ReaxFF-lg models of nitromethane that predict a variety of material properties over a wide range of thermodynamic states are obtained by screening a library of ∼6600 potentials that were previously optimized through the Multiple Objective Evolutionary Strategies (MOES) approach using a training set that included information for other energetic materials composed of carbon, hydrogen, nitrogen, and oxygen. Models that best match experimental nitromethane lattice constants at 4.2 K and 1 atm are evaluated for transferability to high-pressure states at room temperature and are shown to better predict various liquid- and solid-phase structural, thermodynamic, and transport properties as compared to the existing ReaxFF and ReaxFF-lg parametrizations. Although demonstrated for an energetic material, the library of ReaxFF-lg models is supplied to the scientific community to enable new research explorations of complex reactive phenomena in a variety of materials research applications. read less
NOT USED (low confidence) Y. Long and J. Chen, “Theoretical Study of the Interfacial Force-Field, Thermodynamic Property, and Heat Stress for Plastic Bonded Explosives,” Journal of Physical Chemistry C. 2017. link Times cited: 12
Abstract: The force-fields across the TATB/(paraffin, fluoropolymer), … read more
Abstract: The force-fields across the TATB/(paraffin, fluoropolymer), RDX/TATB, RDX/graphite and fluoropolymer/graphite interfaces are obtained by first-principles calculations and parameter optimization. Based on them, the composite materials are simulated in atomistic scale, and a set of thermodynamic properties are calculated, including the heat capacity, thermal expansion coefficient, Gruneisen coefficient, isothermal curve, Hugoniot curve, pressure field, and tension field. We find that the thermal expansion coefficient difference across the explosive/additive interface induces interfacial tension in warming process, the interfacial tension induces positive pressure on the explosive particle, and the positive pressure restrains the thermal expansion of the composite material. A physical picture to describe the influence mechanism of the interface effect on the composite property is obtained. read less
NOT USED (low confidence) C. M. Ashraf and A. V. van Duin, “Extension of the ReaxFF Combustion Force Field toward Syngas Combustion and Initial Oxidation Kinetics.,” The journal of physical chemistry. A. 2017. link Times cited: 170
Abstract: A detailed insight of key reactive events related to oxidati… read more
Abstract: A detailed insight of key reactive events related to oxidation and pyrolysis of hydrocarbon fuels further enhances our understanding of combustion chemistry. Though comprehensive kinetic models are available for smaller hydrocarbons (typically C3 or lower), developing and validating reaction mechanisms for larger hydrocarbons is a daunting task, due to the complexity of their reaction networks. The ReaxFF method provides an attractive computational method to obtain reaction kinetics for complex fuel and fuel mixtures, providing an accuracy approaching ab-initio-based methods but with a significantly lower computational expense. The development of the first ReaxFF combustion force field by Chenoweth et al. (CHO-2008 parameter set) in 2008 has opened new avenues for researchers to investigate combustion chemistry from the atomistic level. In this article, we seek to address two issues with the CHO-2008 ReaxFF description. While the CHO-2008 description has achieved significant popularity for studying large hydrocarbon combustion, it fails to accurately describe the chemistry of small hydrocarbon oxidation, especially conversion of CO2 from CO, which is highly relevant to syngas combustion. Additionally, the CHO-2008 description was obtained faster than expected H abstraction by O2 from hydrocarbons, thus underestimating the oxidation initiation temperature. In this study, we seek to systemically improve the CHO-2008 description and validate it for these cases. Additionally, our aim was to retain the accuracy of the 2008 description for larger hydrocarbons and provide similar quality results. Thus, we expanded the ReaxFF CHO-2008 DFT-based training set by including reactions and transition state structures relevant to the syngas and oxidation initiation pathways and retrained the parameters. To validate the quality of our force field, we performed high-temperature NVT-MD simulations to study oxidation and pyrolysis of four different hydrocarbon fuels, namely, syngas, methane, JP-10, and n-butylbenzene. Results obtained from syngas and methane oxidation simulation indicated that our redeveloped parameters (named as the CHO-2016 parameter set) has significantly improved the C1 chemistry predicted by ReaxFF and has solved the low-temperature oxidation initiation problem. Moreover, Arrhenius parameters of JP-10 decomposition and initiation mechanism pathways of n-butylbenzene pyrolysis obtained using the CHO-2016 parameter set are also in good agreement with both experimental and CHO-2008 simulation results. This demonstrated the transferability of the CHO-2016 description for a wide range of hydrocarbon chemistry. read less
NOT USED (low confidence) L.-lin Liu, P. Liu, S. Hu, and G. He, “Ab Initio Calculations of the N-N Bond Dissociation for the Gas-phase RDX and HMX,” Scientific Reports. 2017. link Times cited: 11
NOT USED (low confidence) Y. Li, R. Kalia, A. Nakano, and P. Vashishta, “Multistage reaction pathways in detonating RDX.” 2017. link Times cited: 7
Abstract: Atomistic mechanisms underlying the reaction time and interm… read more
Abstract: Atomistic mechanisms underlying the reaction time and intermediate reaction products of detonating high explosives far from equilibrium have been elusive. This is because detonation is one of the hardest multiscale physics problems, in which diverse length and time scales play important roles. Here, large spatiotemporal-scale reactive molecular dynamics simulations validated by quantum molecular dynamics simulations reveal a two-stage reaction mechanism during the detonation of cyclotrimethylenetrinitramine cystal. Rapid production of N2 and H2O within ∼10 ps is followed by delayed production of CO molecules beyond ns. We found that further decomposition towards the final products is inhibited by the formation of large metastable carbon- and oxygen- rich clusters with fractal geometry. In addition, we found distinct unimolecular and intermolecular reaction pathways, respectively, for the rapid N2 and H2O productions. read less
NOT USED (low confidence) Y. Li et al., “Anisotropic mechanoresponse of energetic crystallites: a quantum molecular dynamics study of nano-collision.,” Nanoscale. 2016. link Times cited: 1
Abstract: At the nanoscale, chemistry can happen quite differently due… read more
Abstract: At the nanoscale, chemistry can happen quite differently due to mechanical forces selectively breaking the chemical bonds of materials. The interaction between chemistry and mechanical forces can be classified as mechanochemistry. An example of archetypal mechanochemistry occurs at the nanoscale in anisotropic detonating of a broad class of layered energetic molecular crystals bonded by inter-layer van der Waals (vdW) interactions. Here, we introduce an ab initio study of the collision, in which quantum molecular dynamic simulations of binary collisions between energetic vdW crystallites, TATB molecules, reveal atomistic mechanisms of anisotropic shock sensitivity. The highly sensitive lateral collision was found to originate from the twisting and bending to breaking of nitro-groups mediated by strong intra-layer hydrogen bonds. This causes the closing of the electronic energy gap due to an inverse Jahn-Teller effect. On the other hand, the insensitive collisions normal to multilayers are accomplished by more delocalized molecular deformations mediated by inter-layer interactions. Our nano-collision studies provide a much needed atomistic understanding for the rational design of insensitive energetic nanomaterials and the detonation synthesis of novel nanomaterials. read less
NOT USED (low confidence) D. Furman, F. Dubnikova, A. Duin, Y. Zeiri, and R. Kosloff, “Reactive Force Field for Liquid Hydrazoic Acid with Applications to Detonation Chemistry,” Journal of Physical Chemistry C. 2016. link Times cited: 21
Abstract: The development of a reactive force field (ReaxFF formalism)… read more
Abstract: The development of a reactive force field (ReaxFF formalism) for hydrazoic acid (HN3), a highly sensitive liquid energetic material, is reported. The force field accurately reproduces results of density functional theory (DFT) calculations. The quality and performance of the force field are examined by detailed comparison with DFT calculations related to uni, bi, and trimolecular thermal decomposition routes. Reactive molecular dynamics (RMD) simulations are performed to reveal the initial chemical events governing the detonation chemistry of liquid HN3. The outcome of these simulations compares very well with recent results of tight-binding DFT molecular dynamics and thermodynamic calculations. On the basis of our RMD simulations, predictions were made for the activation energies and volumes in a broad range of temperatures and initial material compressions. read less
NOT USED (low confidence) K. L. Joshi, M. Losada, and S. Chaudhuri, “Intermolecular Energy Transfer Dynamics at a Hot-Spot Interface in RDX Crystals.,” The journal of physical chemistry. A. 2016. link Times cited: 18
Abstract: The phonon mediated vibrational up-pumping mechanisms assume… read more
Abstract: The phonon mediated vibrational up-pumping mechanisms assume an intact lattice and climbing of a vibrational ladder using strongly correlated multiphonon dynamics under equilibrium or near-equilibrium conditions. Important dynamic processes far from-equilibrium in regions of large temperature gradient after the onset of decomposition reactions in energetic solids are relatively unknown. In this work, we present a classical molecular dynamics (MD) simulation-based study of such processes using a nonreactive and a reactive potential to study a fully reacted and unreacted zone in RDX (1,3,5-trinitro-1,3,5-triazocyclohexane) crystal under nonequilibrium conditions. The energy transfer rate is evaluated as a function of temperature difference between the reacted and unreacted regions, and for different widths and cross-sectional area of unreacted RDX layers. Vibrational up-pumping processes probed using velocity autocorrelation functions indicate that the mechanisms at high-temperature interfaces are quite different from the standard phonon-based models proposed in current literature. In particular, the up-pumping of high-frequency vibrations are seen in the presence of small molecule collisions at the hot-spot interface with strong contributions from bending modes. It also explains some major difference in the order of decomposition of C-N and N-N bonds as seen in recent literature on initiation chemistry. read less
NOT USED (low confidence) J. Yuan, G. Ji, X.-R. Chen, D. Wei, F. Zhao, and Q. Wu, “Phase transition, thermodynamics properties and IR spectrum of α- and γ-RDX: First principles and MD studies,” Chemical Physics Letters. 2016. link Times cited: 10
NOT USED (low confidence) D. Guo, S. Zybin, Q. An, W. Goddard, and F. Huang, “Prediction of the Chapman-Jouguet chemical equilibrium state in a detonation wave from first principles based reactive molecular dynamics.,” Physical chemistry chemical physics : PCCP. 2016. link Times cited: 22
Abstract: The combustion or detonation of reacting materials at high t… read more
Abstract: The combustion or detonation of reacting materials at high temperature and pressure can be characterized by the Chapman-Jouguet (CJ) state that describes the chemical equilibrium of the products at the end of the reaction zone of the detonation wave for sustained detonation. This provides the critical properties and product kinetics for input to macroscale continuum simulations of energetic materials. We propose the ReaxFF Reactive Dynamics to CJ point protocol (Rx2CJ) for predicting the CJ state parameters, providing the means to predict the performance of new materials prior to synthesis and characterization, allowing the simulation based design to be done in silico. Our Rx2CJ method is based on atomistic reactive molecular dynamics (RMD) using the QM-derived ReaxFF force field. We validate this method here by predicting the CJ point and detonation products for three typical energetic materials. We find good agreement between the predicted and experimental detonation velocities, indicating that this method can reliably predict the CJ state using modest levels of computation. read less
NOT USED (low confidence) A. Rahnamoun and A. Duin, “Study of thermal conductivity of ice clusters after impact deposition on the silica surfaces using the ReaxFF reactive force field.,” Physical chemistry chemical physics : PCCP. 2016. link Times cited: 5
Abstract: During aircraft or spacecraft missions, ice accumulates on d… read more
Abstract: During aircraft or spacecraft missions, ice accumulates on different parts of their surface elements. An important parameter affecting the ability to remove this ice from the surface is the heat transfer characteristics of the accumulated ice. The ice heat transfer is related to the process of ice formation and its density and internal structure. In this study we investigate the effects of the ice and silica structure and the ice cluster attachment mechanism to the silica surface on the thermal conductivity (TC) of the attached ice cluster using the ReaxFF reactive force field. The purpose of this study is to investigate the thermal transport in amorphous and crystalline ice after high-velocity deposition on the silica surfaces. A dual thermostat method has been applied for the calculation of TC values. The validity of this method has been verified by comparing the calculated values of TC for crystal and amorphous ice with available experimental values. Our calculations show that the TC values of both crystal and amorphous ice drop after deposition on the silica surfaces. This decrease in the TC is more significant for the ice deposition on suboxide silica surfaces. Furthermore, crystal ice shows higher TC values than amorphous ice after accumulation. However, when crystal ice impacts on the silica surface at 1 km s(-1) impact speed, the crystalline shape of the ice cluster is lost to a considerable level and the TC values obtained for the ice clusters in such cases are closer to amorphous ice TC values. We observed a decrease in the TC values when ionic species are added inside the ice clusters. read less
NOT USED (low confidence) W. Zhang and A. Duin, “ReaxFF Reactive Molecular Dynamics Simulation of Functionalized Poly(phenylene oxide) Anion Exchange Membrane,” Journal of Physical Chemistry C. 2015. link Times cited: 60
Abstract: Three functionalized poly(phenylene oxide) (PPO) anion excha… read more
Abstract: Three functionalized poly(phenylene oxide) (PPO) anion exchange membranes (AEMs), PPO–trimethylamine (PPO–TMA), PPO–dimethylbutylamine (PPO–DMBA), and PPO–dimethyloctylamine (PPO–DMOA), at two hydration levels (λ = 8.3 and 20.8) have been studied by ReaxFF reactive molecular dynamics simulations. Our simulations reveal that with increasing hydration the microstructures of membrane swell and water molecules are more likely to form a channel, which improves the diffusion of hydroxide ion (OH–). Our study of OH– diffusion demonstrates that PPO–TMA hydrated membrane provides the biggest diffusion constant at the high hydration level. However, from comparison of the structural and dynamical properties of the three membranes at the same water content, it is found that when one methyl group of quaternary ammonium center is replaced by a long alkyl chain group, the hydrophobic effects block the OH– approaching nitrogen, resulting in a lower rate of degradation and an improved alkaline stability of PPO–DMOA hydrat... read less
NOT USED (low confidence) D. Yilmaz, “Modeling failure mechanisms of poly(p-phenylene terephthalamide) fiber using reactive potentials,” Computational Materials Science. 2015. link Times cited: 21
NOT USED (low confidence) B. Saha, A. Furmanchuk, Y. Dzenis, and G. Schatz, “Multi-step mechanism of carbonization in templated polyacrylonitrile derived fibers: ReaxFF model uncovers origins of graphite alignment,” Carbon. 2015. link Times cited: 48
NOT USED (low confidence) L. C. Lin, J. Choi, and J. Grossman, “Two-dimensional covalent triazine framework as an ultrathin-film nanoporous membrane for desalination.,” Chemical communications. 2015. link Times cited: 89
Abstract: We computationally demonstrate that two-dimensional covalent… read more
Abstract: We computationally demonstrate that two-dimensional covalent triazine frameworks (CTFs) provide opportunities in water desalination. By varying the chemical building blocks, the pore structure, chemistry, and membrane performance can be designed, leading to two orders of magnitude higher water permeability than polyamide membranes while maintaining excellent ability to reject salts. read less
NOT USED (low confidence) M. Wood, M. Cherukara, E. Kober, and A. Strachan, “Ultrafast Chemistry under Nonequilibrium Conditions and the Shock to Deflagration Transition at the Nanoscale,” Journal of Physical Chemistry C. 2015. link Times cited: 103
Abstract: We use molecular dynamics simulations to describe the chemic… read more
Abstract: We use molecular dynamics simulations to describe the chemical reactions following shock-induced collapse of cylindrical pores in the high-energy density material RDX. For shocks with particle velocities of 2 km/s we find that the collapse of a 40 nm diameter pore leads to a deflagration wave. Molecular collisions during the collapse lead to ultrafast, multistep chemical reactions that occur under nonequilibrium conditions. Exothermic products formed during these first few picoseconds prevent the nanoscale hotspot from quenching. Within 30 ps, a local deflagration wave develops; it propagates at 0.25 km/s and consists of an ultrathin reaction zone of only ∼5 nm, thus involving large temperature and composition gradients. Contrary to the assumptions in current models, a static thermal hotspot matching the dynamical one in size and thermodynamic conditions fails to produce a deflagration wave indicating the importance of nonequilibrium loading in the criticality of nanoscale hot spots. These results provide... read less
NOT USED (low confidence) Y. Thakur et al., “Optimizing nanostructure to achieve high dielectric response with low loss in strongly dipolar polymers,” Nano Energy. 2015. link Times cited: 41
NOT USED (low confidence) Q.-L. Yan, S. Zeman, X.-H. Zhang, J. Málek, and W.-xi Xie, “The mechanisms for desensitization effect of synthetic polymers on BCHMX: Physical models and decomposition pathways.,” Journal of hazardous materials. 2015. link Times cited: 10
NOT USED (low confidence) B. Chen, Z.-J. Diao, Y. Zhao, and X.-xun Ma, “A ReaxFF molecular dynamics (MD) simulation for the hydrogenation reaction with coal related model compounds,” Fuel. 2015. link Times cited: 22
NOT USED (low confidence) S. Naserifar, W. Goddard, T. Tsotsis, and M. Sahimi, “First principles-based multiparadigm, multiscale strategy for simulating complex materials processes with applications to amorphous SiC films.,” The Journal of chemical physics. 2015. link Times cited: 9
Abstract: Progress has recently been made in developing reactive force… read more
Abstract: Progress has recently been made in developing reactive force fields to describe chemical reactions in systems too large for quantum mechanical (QM) methods. In particular, ReaxFF, a force field with parameters that are obtained solely from fitting QM reaction data, has been used to predict structures and properties of many materials. Important applications require, however, determination of the final structures produced by such complex processes as chemical vapor deposition, atomic layer deposition, and formation of ceramic films by pyrolysis of polymers. This requires the force field to properly describe the formation of other products of the process, in addition to yielding the final structure of the material. We describe a strategy for accomplishing this and present an example of its use for forming amorphous SiC films that have a wide variety of applications. Extensive reactive molecular dynamics (MD) simulations have been carried out to simulate the pyrolysis of hydridopolycarbosilane. The reaction products all agree with the experimental data. After removing the reaction products, the system is cooled down to room temperature at which it produces amorphous SiC film, for which the computed radial distribution function, x-ray diffraction pattern, and the equation of state describing the three main SiC polytypes agree with the data and with the QM calculations. Extensive MD simulations have also been carried out to compute other structural properties, as well the effective diffusivities of light gases in the amorphous SiC film. read less
NOT USED (low confidence) Y. Wen, C. Zhang, X. Xue, and X. Long, “Cluster evolution during the early stages of heating explosives and its relationship to sensitivity: a comparative study of TATB, β-HMX and PETN by molecular reactive force field simulations.,” Physical chemistry chemical physics : PCCP. 2015. link Times cited: 43
Abstract: Clustering is experimentally and theoretically verified duri… read more
Abstract: Clustering is experimentally and theoretically verified during the complicated processes involved in heating high explosives, and has been thought to influence their detonation properties. However, a detailed description of the clustering that occurs has not been fully elucidated. We used molecular dynamic simulations with an improved reactive force field, ReaxFF_lg, to carry out a comparative study of cluster evolution during the early stages of heating for three representative explosives: 1,3,5-triamino-2,4,6-trinitrobenzene (TATB), β-1,3,5,7-tetranitro-1,3,5,7-tetrazocine (HMX) and pentaerythritol tetranitrate (PETN). These representatives vary greatly in their oxygen balance (OB), molecular structure, stability and experimental sensitivity. We found that when heated, TATB, HMX and PETN differ in the size, amount, proportion and lifetime of their clusters. We also found that the clustering tendency of explosives decreases as their OB becomes less negative. We propose that the relationship between OB and clustering can be attributed to the role of clustering in detonation. That is, clusters can form more readily in a high explosive with a more negative OB, which retard its energy release, secondary decomposition, further decomposition to final small molecule products and widen its detonation reaction zone. Moreover, we found that the carbon content of the clusters increases during clustering, in accordance with the observed soot, which is mainly composed of carbon as the final product of detonation or deflagration. read less
NOT USED (low confidence) Y. Long and J. Chen, “Systematic study of the reaction kinetics for HMX.,” The journal of physical chemistry. A. 2015. link Times cited: 29
Abstract: The reaction process of octahydro-1,3,5,7-tetranitro-1,3,5,7… read more
Abstract: The reaction process of octahydro-1,3,5,7-tetranitro-1,3,5,7-tetrazocine (HMX) in wide temperature and pressure ranges is simulated by molecular dynamics. A set of postprocessing programs is written to evaluate the intermediate molecules and chemical reactions. On the basis of these evaluations, the reaction rates, reactive Hugoniot curves, and detonation wave profile are calculated. The detonation velocity and detonation pressure are determined as 9984 m/s and 38.3349 GPa, in agreement with the experimental results, 9110 m/s and 39.5 GPa. The width of the reaction zone is 10 μm, and the main products are N2, H2O, and CO2. We find some molecules play an important role in intermediate reactions but are not exhibited in final products, such as N2O2, N2O5, and C3H3N3. read less
NOT USED (low confidence) T. Zhou, J. Lou, H. Song, and F. Huang, “Anisotropic shock sensitivity in a single crystal δ-cyclotetramethylene tetranitramine: a reactive molecular dynamics study.,” Physical chemistry chemical physics : PCCP. 2015. link Times cited: 9
Abstract: The anisotropic shock sensitivity in a single crystal δ-cycl… read more
Abstract: The anisotropic shock sensitivity in a single crystal δ-cyclotetramethylene tetranitramine (δ-HMX) was investigated using the compress-shear reactive dynamics (CS-RD) computational protocol. Significant anisotropies in the thermo-mechanical and chemical responses were found by measuring the shear stress, energy, temperature, and chemical reactions during the dynamical process for the shock directions perpendicular to the (100), (010), (001), (110), (101), (011), and (111) planes. We predict that δ-HMX is sensitive for the shocks perpendicular to the (111), (011), (110), and (101) planes, which is intermediate to the (100) and (010) plane and is insensitive to the (001) plane. The internal energy accumulated within the duration of the surmounting shear stress barrier is a useful criterion to distinguish the sensitive directions from the less sensitive ones. The molecular origin of the anisotropic sensitivity is suggested to be the intermolecular steric arrangements across a slip plane induced by shock compression. The shear deformation induced by the shock along the sensitive direction encounters strong intermolecular contacts and has small intermolecular free space for geometry relaxation when the molecules collide, leading to high shear stress barriers and energy accumulation, which benefits the temperature increase and initial chemical bond breaking that trigger further reactions. read less
NOT USED (low confidence) D. Guo, Q. An, S. Zybin, W. Goddard, F. Huang, and B. Tang, “The co-crystal of TNT/CL-20 leads to decreased sensitivity toward thermal decomposition from first principles based reactive molecular dynamics,” Journal of Materials Chemistry. 2015. link Times cited: 63
Abstract: To gain an atomistic-level understanding of the experimental… read more
Abstract: To gain an atomistic-level understanding of the experimental observation that the cocrystal TNT/CL-20 leads to decreased sensitivity, we carried out reactive molecular dynamics (RMD) simulations using the ReaxFF reactive force field. We compared the thermal decomposition of the TNT/CL-20 cocrystal with that of pure crystals of TNT and CL-20 and with a simple physical mixture of TNT and CL-20. We find that cocrystal has a lower decomposition rate than CL-20 but higher than TNT, which is consistent with experimental observation. We find that the formation of carbon clusters arising from TNT, a carbon-rich molecule, plays an important role in the thermal decomposition process, explaining the decrease in sensitivity for the cocrystal. At low temperature and in the early stage of chemical reactions under high temperature, the cocrystal releases energy more slowly than the simple mixture of CL-20–TNT. These results confirm the expectation that co-crystallization is an effective way to decrease the sensitivity for energetic materials while retaining high performance. read less
NOT USED (low confidence) P. Politzer and J. Murray, “Some molecular/crystalline factors that affect the sensitivities of energetic materials: molecular surface electrostatic potentials, lattice free space and maximum heat of detonation per unit volume,” Journal of Molecular Modeling. 2015. link Times cited: 111
NOT USED (low confidence) C. Ye et al., “Initial decomposition reaction of di-tetrazine-tetroxide (DTTO) from quantum molecular dynamics: implications for a promising energetic material,” Journal of Materials Chemistry. 2015. link Times cited: 29
Abstract: Di-tetrazine-tetroxide (DTTO) was predicted in 2001 to have … read more
Abstract: Di-tetrazine-tetroxide (DTTO) was predicted in 2001 to have a density (up to 2.3 g cm^(−3)) and heat of detonation (up to 421.0 kcal mol^(−1)) better than other explosives, making it the “holy grail” of energetic materials (EMs), but all attempts at synthesis have failed. We report Density Functional Theory (DFT) molecular dynamics simulations (DFT-MD) on DTTO crystal for the two most stable monomers. We predict that the most stable isomer (c1) has a density of ρ = 1.96 g cm^(−3) with an estimated detonation velocity (D_v) of 9.70 km s^(−1) and a detonation pressure (D_p) of 43.0 GPa, making it comparable to RDX (ρ = 1.82 g cm^(−3), D_v = 8.75 km s^(−1), D_p = 35.0 GPa), HMX (ρ = 1.91 g cm^(−3), D_v = 9.10 km s^(−1), D_p = 39.3 GPa) and CL-20 (ρ = 2.04 g cm^(−3), D_v = 9.38 km s^(−1), D_p = 44.1 GPa). The DFT-MD studies find that the initial reaction at lower pressure is unimolecular decomposition to form two N_2O molecules (barrier 45.9 kcal mol^(−1)), while at higher pressure it is intermolecular oxygen-transfer with a barrier of 40.1 kcal mol^(−1). For the c2 isomer (less stable by 1.2 kcal mol^(−1)) the initial reaction involves two DTTO molecules reacting to form a dimer which then releases N_2 as a direct product (barrier 48.1 kcal mol^(−1)), a unique initial reaction among EMs. These results suggest that DTTO may have a higher thermal stability (barrier >7.0 kcal mol^(−1) higher) than RDX, HMX, and CL-20. read less
NOT USED (low confidence) D.-C. Yue et al., “Tribochemical mechanism of amorphous silica asperities in aqueous environment: a reactive molecular dynamics study.,” Langmuir : the ACS journal of surfaces and colloids. 2015. link Times cited: 49
Abstract: Reactive molecular dynamics (ReaxFF) simulations are used to… read more
Abstract: Reactive molecular dynamics (ReaxFF) simulations are used to explore the atomic-level tribochemical mechanism of amorphous silica (a-SiO2) in a nanoscale, single-asperity contact in an aqueous environment. These sliding simulations are performed in both a phosphoric acid solution and in pure water under different normal pressures. The results show that tribochemical processes have profound consequences on tribological performance. Water molecules could help avoid direct adhesive interaction between a-SiO2 surfaces in pure water under low normal load. However, formation and rupture of interfacial siloxane bonds are obviously observed under higher normal load. In phosphoric acid solution, polymerization of phosphoric acid molecules occurs, yielding oligomers under lower load, and tribochemical reactions between the molecules and the sliding surfaces could enhance wear under higher load. The bridging oxygen atoms in silica play an important role in the formation of interfacial covalent bonds, and hydrogen is found to have a weakening effect on these bonds, resulting in the rupture during shear-related loading. This work sheds light on tribochemical reactions as a mechanism for lubrication and wear in water-based or other tribological systems. read less
NOT USED (low confidence) K. S. Rao, F. Yehya, A. Chaudhary, A. S. Kumar, and A. Sahoo, “Thermal stability study of nitro-rich triazole derivatives using temperature dependent time resolved pulsed photoacoustic (PA) technique,” Journal of Analytical and Applied Pyrolysis. 2014. link Times cited: 20
NOT USED (low confidence) Q. An, W. Goddard, S. Zybin, and S. Luo, “Inhibition of Hotspot Formation in Polymer Bonded Explosives Using an Interface Matching Low Density Polymer Coating at the Polymer–Explosive Interface,” Journal of Physical Chemistry C. 2014. link Times cited: 21
Abstract: In order to elucidate how shocks in heterogeneous materials … read more
Abstract: In order to elucidate how shocks in heterogeneous materials affect decomposition and reactive processes, we used the ReaxFF reactive force field in reactive molecules dynamics (RMD) simulations of the effects of strong shocks (2.5 and 3.5 km/s) on a prototype polymer bonded explosive (PBX) consisting of cyclotrimethylene trinitramine (RDX) bonded to hydroxyl-terminated polybutadiene (HTPB). We showed earlier that shock propagation from the high density RDX to the low density polymer (RDX → Poly) across a nonplanar periodic interface (sawtooth) leads to a hotspot at the initial asperity but no additional hotspot at the second asperity. This hotspot arises from shear along the interface induced by relaxation of the stress at the asperity. We now report the case for shock propagation from the low density polymer to the high density RDX (Poly → RDX) where we find a hotspot at the initial asperity and a second more dramatic hotspot at the second asperity. This second hotspot is enhanced due to shock wave conve... read less
NOT USED (low confidence) C. Zhang, Y. Wen, and X. Xue, “Self-enhanced catalytic activities of functionalized graphene sheets in the combustion of nitromethane: molecular dynamic simulations by molecular reactive force field.,” ACS applied materials & interfaces. 2014. link Times cited: 41
Abstract: Functionalized graphene sheet (FGS) is a promising additive … read more
Abstract: Functionalized graphene sheet (FGS) is a promising additive that enhances fuel/propellant combustion, and the determination of its mechanism has attracted much interest. In the present study, a series of molecular dynamic simulations based on a reactive force field (ReaxFF) are performed to explore the catalytic activity (CA) of FGS in the thermal decay of nitromethane (NM, CH3NO2). FGSs and pristine graphene sheets (GSs) are oxidized in hot NM liquid to increase their functionalities and subsequently show self-enhanced CAs during the decay. The CAs result from the interatomic exchanges between the functional groups on the sheets and the NM liquid, i.e., mainly between H and O atoms. CA is dependent on the density of NM, functionalities of sheets, and temperature. The GSs and FGSs that originally exhibit different functionalities tend to possess similar functionalities and consequently similar CAs as temperature increases. Other carbon materials and their oxides can accelerate combustion of other fuels/propellants similar to NM, provided that they can be dispersed and their key reaction steps in combustion are similar to NM. read less
NOT USED (low confidence) T.-R. Shan and A. Thompson, “Shock-induced hotspot formation and chemical reaction initiation in PETN containing a spherical void,” Journal of Physics: Conference Series. 2014. link Times cited: 31
Abstract: We present results of reactive molecular dynamics simulation… read more
Abstract: We present results of reactive molecular dynamics simulations of hotspot formation and chemical reaction initiation in shock-induced compression of pentaerythritol tetranitrate (PETN) with the ReaxFF reactive force field. A supported shockwave is driven through a PETN crystal containing a 20 nm spherical void at a sub-threshold impact velocity of 2 km/s. Formation of a hotspot due to shock-induced void collapse is observed. During void collapse, NO2 is the dominant species ejected from the upstream void surface. Once the ejecta collide with the downstream void surface and the hotspot develops, formation of final products such as N2 and H2O is observed. The simulation provides a detailed picture of how void collapse and hotspot formation leads to initiation at sub-threshold impact velocities. read less
NOT USED (low confidence) A. Rahnamoun and A. V. van Duin, “Reactive molecular dynamics simulation on the disintegration of Kapton, POSS polyimide, amorphous silica, and teflon during atomic oxygen impact using the ReaxFF reactive force-field method.,” The journal of physical chemistry. A. 2014. link Times cited: 82
Abstract: Atomic oxygen (AO) is the most abundant element in the low E… read more
Abstract: Atomic oxygen (AO) is the most abundant element in the low Earth orbit (LEO). It is the result of the dissociation of molecular oxygen by ultraviolet radiation from the sun. In the LEO, it collides with the materials used on spacecraft surfaces and causes degradation of these materials. The degradation of the materials on the surface of spacecrafts at LEO has been a significant problem for a long time. Kapton polyimide, polyhedral oligomeric silsesquioxane (POSS), silica, and Teflon are the materials extensively used in spacecraft industry, and like many other materials used in spacecraft industry, AO collision degradation is an important issue in their applications on spacecrafts. To investigate the surface chemistry of these materials in exposure to space AO, a computational chemical evaluation of the Kapton polyimide, POSS, amorphous silica, and Teflon was performed in separate simulations under similar conditions. For performing these simulations, the ReaxFF reactive force-field program was used, which provides the computational speed required to perform molecular dynamics (MD) simulations on system sizes sufficiently large to describe the full chemistry of the reactions. Using these simulations, the effects of AO impact on different materials and the role of impact energies, the content of material, and temperature of material on the behavior of the materials are studied. The ReaxFF results indicate that Kapton is less resistant than Teflon toward AO damage. These results are in good agreement with experiment. These simulations indicate that the amorphous silica shows the highest stability among these materials before the start of the highly exothermic silicon oxidation. We have verified that adding silicon to the bulk of the Kapton structure enhances the stability of the Kapton against AO impact. Our canonical MD simulations demonstrate that an increase in the heat transfer in materials during AO impact can provide a considerable decrease in the disintegration of the material. This effect is especially relevant in silica AO collision. Considerable experimental efforts have been undertaken to minimize such AO-based degradations. As our simulations demonstrate, ReaxFF can provide a cost-effective screening tool for future material optimization. read less
NOT USED (low confidence) K.-H. Lin, B. Holian, T. Germann, and A. Strachan, “Mesodynamics with implicit degrees of freedom.,” The Journal of chemical physics. 2014. link Times cited: 19
Abstract: Mesoscale phenomena--involving a level of description betwee… read more
Abstract: Mesoscale phenomena--involving a level of description between the finest atomistic scale and the macroscopic continuum--can be studied by a variation on the usual atomistic-level molecular dynamics (MD) simulation technique. In mesodynamics, the mass points, rather than being atoms, are mesoscopic in size, for instance, representing the centers of mass of polycrystalline grains or molecules. In order to reproduce many of the overall features of fully atomistic MD, which is inherently more expensive, the equations of motion in mesodynamics must be derivable from an interaction potential that is faithful to the compressive equation of state, as well as to tensile de-cohesion that occurs along the boundaries of the mesoscale units. Moreover, mesodynamics differs from Newton's equations of motion in that dissipation--the exchange of energy between mesoparticles and their internal degrees of freedom (DoFs)--must be described, and so should the transfer of energy between the internal modes of neighboring mesoparticles. We present a formulation where energy transfer between the internal modes of a mesoparticle and its external center-of-mass DoFs occurs in the phase space of mesoparticle coordinates, rather than momenta, resulting in a Galilean invariant formulation that conserves total linear momentum and energy (including the energy internal to the mesoparticles). We show that this approach can be used to describe, in addition to mesoscale problems, conduction electrons in atomic-level simulations of metals, and we demonstrate applications of mesodynamics to shockwave propagation and thermal transport. read less
NOT USED (low confidence) D. Furman et al., “Decomposition of condensed phase energetic materials: interplay between uni- and bimolecular mechanisms.,” Journal of the American Chemical Society. 2014. link Times cited: 116
Abstract: Activation energy for the decomposition of explosives is a c… read more
Abstract: Activation energy for the decomposition of explosives is a crucial parameter of performance. The dramatic suppression of activation energy in condensed phase decomposition of nitroaromatic explosives has been an unresolved issue for over a decade. We rationalize the reduction in activation energy as a result of a mechanistic change from unimolecular decomposition in the gas phase to a series of radical bimolecular reactions in the condensed phase. This is in contrast to other classes of explosives, such as nitramines and nitrate esters, whose decomposition proceeds via unimolecular reactions both in the gas and in the condensed phase. The thermal decomposition of a model nitroaromatic explosive, 2,4,6-trinitrotoluene (TNT), is presented as a prime example. Electronic structure and reactive molecular dynamics (ReaxFF-lg) calculations enable to directly probe the condensed phase chemistry under extreme conditions of temperature and pressure, identifying the key bimolecular radical reactions responsible for the low activation route. This study elucidates the origin of the difference between the activation energies in the gas phase (~62 kcal/mol) and the condensed phase (~35 kcal/mol) of TNT and identifies the corresponding universal principle. On the basis of these findings, the different reactivities of nitro-based organic explosives are rationalized as an interplay between uni- and bimolecular processes. read less
NOT USED (low confidence) F. Guo, H. Zhang, H. Hu, and X. Cheng, “Effects of hydrogen bonds on solid state TATB, RDX, and DATB under high pressures,” Chinese Physics B. 2014. link Times cited: 15
Abstract: To probe the behavior of hydrogen bonds in solid energetic m… read more
Abstract: To probe the behavior of hydrogen bonds in solid energetic materials, we conduct ReaxFF and SCC–DFTB molecular dynamics simulations of crystalline TATB, RDX, and DATB. By comparing the intra- and inter-molecular hydrogen bonding rates, we find that the crystal structures are stabilized by inter-molecular hydrogen bond networks. Under high-pressure, the inter- and intra-molecular hydrogen bonds in solid TATB and DATB are nearly equivalent. The hydrogen bonds in solid TATB and DATB are much shorter than in solid RDX, which suggests strong hydrogen bond interactions existing in these energetic materials. Stretching of the C–H bond is observed in solid RDX, which may lead to further decomposition and even detonation. read less
NOT USED (low confidence) T.-R. Shan, A. V. van Duin, and A. Thompson, “Development of a ReaxFF reactive force field for ammonium nitrate and application to shock compression and thermal decomposition.,” The journal of physical chemistry. A. 2014. link Times cited: 28
Abstract: We have developed a new ReaxFF reactive force field parametr… read more
Abstract: We have developed a new ReaxFF reactive force field parametrization for ammonium nitrate. Starting with an existing nitramine/TATB ReaxFF parametrization, we optimized it to reproduce electronic structure calculations for dissociation barriers, heats of formation, and crystal structure properties of ammonium nitrate phases. We have used it to predict the isothermal pressure-volume curve and the unreacted principal Hugoniot states. The predicted isothermal pressure-volume curve for phase IV solid ammonium nitrate agreed with electronic structure calculations and experimental data within 10% error for the considered range of compression. The predicted unreacted principal Hugoniot states were approximately 17% stiffer than experimental measurements. We then simulated thermal decomposition during heating to 2500 K. Thermal decomposition pathways agreed with experimental findings. read less
NOT USED (low confidence) M. Wood, A. V. van Duin, and A. Strachan, “Coupled thermal and electromagnetic induced decomposition in the molecular explosive αHMX; a reactive molecular dynamics study.,” The journal of physical chemistry. A. 2014. link Times cited: 103
Abstract: We use molecular dynamics simulations with the reactive pote… read more
Abstract: We use molecular dynamics simulations with the reactive potential ReaxFF to investigate the initial reactions and subsequent decomposition in the high-energy-density material α-HMX excited thermally and via electric fields at various frequencies. We focus on the role of insult type and strength on the energy increase for initial decomposition and onset of exothermic chemistry. We find both of these energies increase with the increasing rate of energy input and plateau as the processes become athermal for high loading rates. We also find that the energy increase required for exothermic reactions and, to a lesser extent, that for initial chemical reactions depend on the insult type. Decomposition can be induced with relatively weak insults if the appropriate modes are targeted but increasing anharmonicities during heating lead to fast energy transfer and equilibration between modes that limit the effect of loading type. read less
NOT USED (low confidence) Q. An, W. Goddard, S. Zybin, A. Jaramillo-Botero, and T. Zhou, “Highly Shocked Polymer Bonded Explosives at a Nonplanar Interface: Hot-Spot Formation Leading to Detonation,” Journal of Physical Chemistry C. 2013. link Times cited: 83
Abstract: We report reactive molecular dynamics simulations using the … read more
Abstract: We report reactive molecular dynamics simulations using the ReaxFF reactive force field to examine shock-induced hot-spot formation followed by detonation initiation in realistic (2.7 million atoms) models of polymer bonded explosives (PBX) with nonplanar interfaces. We considered here two energetic materials (EMs) pentaerythritol tetranitrate (PETN), a common EM for PBX, and silicon pentaerythritol tetranitrate (Si-PETN), which is so extremely sensitive that it has not been possible to characterize its shock properties experimentally. In each case the EM was embedded in a hydroxyl-terminated polybutadiene (HTPB) based polymer binder matrix to form a model of PBX that has a periodic sawtooth nonplanar interface. For the cases in which the shock wave propagates from the EM to polymer (EM→poly), we observed that a hot spot arises from shear localization at the convex polymer asperity. For the case in which the shock direction is inverted (shock wave propagates from the polymer to the EM, EM←poly), we find t... read less
NOT USED (low confidence) D.-C. Yue et al., “Tribochemistry of Phosphoric Acid Sheared between Quartz Surfaces: A Reactive Molecular Dynamics Study,” Journal of Physical Chemistry C. 2013. link Times cited: 57
Abstract: Tribochemical processes have profound consequences on tribol… read more
Abstract: Tribochemical processes have profound consequences on tribological performance. In the present paper, the tribochemical mechanism of low friction state in the silica/phosphoric acid system is elucidated by reactive molecular dynamics (ReaxFF) simulations. The friction coefficient is found having strong positive correlation with the number of interfacial hydrogen bonds, which suggests that a weaker interfacial hydrogen bond network would favor a lower friction. The friction reduction mechanisms have been analyzed in two temperature regimes: For 300 ≤ T ≤ 600 K, no indication of tribochemical reaction is observed, and the friction coefficient decreases because of the accelerated molecular rotational and translational motion and the corresponding weakened hydrogen bond network. For 800 K ≤ T ≤ 1400 K, the occurrence of tribochemical reactions leads to a clustering and polymerization of the phosphoric acid molecules and generation of a considerable quantity of water molecules distributed mainly in the sliding... read less
NOT USED (low confidence) Z.-J. Diao, Y. Zhao, B. Chen, C. Duan, and S. Song, “ReaxFF reactive force field for molecular dynamics simulations of epoxy resin thermal decomposition with model compound,” Journal of Analytical and Applied Pyrolysis. 2013. link Times cited: 87
NOT USED (low confidence) Y. Cai, F. Zhao, Q. An, H. A. Wu, W. A. Goddard, and S.-N. Luo, “Shock response of single crystal and nanocrystalline pentaerythritol tetranitrate: Implications to hotspot formation in energetic materials.,” The Journal of chemical physics. 2013. link Times cited: 32
Abstract: We investigate shock response of single crystal and nanocrys… read more
Abstract: We investigate shock response of single crystal and nanocrystalline pentaerythritol tetranitrate (PETN) with a coarse-grained model and molecular dynamics simulations, as regards mechanical hotspot formation in the absence or presence of grain boundaries (GBs). Single crystals with different orientations, and columnar nanocrystalline PETN with regular hexagonal, irregular hexagonal, and random GB patterns, are subjected to shock loading at different shock strengths. In single crystals, shock-induced plasticity is consistent with resolved shear stress calculations and the steric hindrance model, and this deformation leads to local heating. For regular-shaped hexagonal columnar nanocrystalline PETN, different misorientation angles lead to activation of different/same slip systems, different deformation in individual grains and as a whole, different GB friction, different temperature distributions, and then, different hotspot characteristics. Compared to their regular-shaped hexagonal counterpart, nanocrystalline PETN with irregular hexagonal GB pattern and that with random GBs, show deformation and hotspot features specific to their GBs. Driven by stress concentration, hotspot formation is directly related to GB friction and GB-initiated crystal plasticity, and the exact deformation is dictated by grain orientations and resolved shear stresses. GB friction alone can induce hotspots, but the hotspot temperature can be enhanced if it is coupled with GB-initiated crystal plasticity, and the slip of GB atoms has components out of the GB plane. The magnitude of shearing can correlate well with temperature, but the slip direction of GB atoms relative to GBs may play a critical role. Wave propagation through varying microstructure may also induce differences in stress states (e.g., stress concentrations) and loading rates, and thus, local temperature rise. GB-related friction and plasticity induce local heating or mechanical hotspots, which could be precursors to chemical hotspot formation related to initiation in energetic materials, in the absence of other, likely more effective, means for hotspot formation such as void collapse. read less
NOT USED (low confidence) C. M. Berg and D. Dlott, “Picosecond dynamics of shock compressed and flash-heated nanometer thick films of δ-HMX,” Journal of Physics: Conference Series. 2013. link Times cited: 5
Abstract: Progress towards probing molecular dynamics of octahydro-1,3… read more
Abstract: Progress towards probing molecular dynamics of octahydro-1,3,5,7-tetranitro-1,3,4,7-tetrazocine (HMX) subjected to shock compression of a few GPa and/or temperature excursions exceeding thermal decomposition values (T > 500 K) is described. Due to shock velocities of a few nm/ps, nanometer-thick layers are needed for picosecond time resolution. Therefore, 5-10 nm thick films of S-HMX were deposited on metallic substrates with a template of a 4-nitrobenzoic acid monolayer. A polymer layer a few microns thick was spin-coated on top of S-HMX for shock confinement. The monolayer and HMX layer were probed simultaneously utilizing an ultrafast nonlinear coherent vibrational spectroscopy, termed vibrational sum-frequency generation (SFG). Shock pressures were estimated via comparisons with the monolayer nitro transition frequency blueshift in hydrostatic pressure measurements. Temperature determinations were made based on the reflectance of the metallic substrate. read less
NOT USED (low confidence) X.-F. Chen, K. Yang, and B.-Z. Wang, “Mechanisms and risk assessments on the N-nitration of N-acetylhexahydro-s-triazines: understanding the preparation of RDX (2).,” The journal of physical chemistry. A. 2013. link Times cited: 3
Abstract: Although the N-nitration by nitric acid is widely used to sy… read more
Abstract: Although the N-nitration by nitric acid is widely used to synthesize nitramines in biological, medical, and explosive industries, little is known about the microscopic behavior when the nitrated substrates are tertiary amines. Hexahydro-1,3,5-triacetyl-s-triazine (TRAT) nitrated into hexahydro-1,3,5-trinitro-s-triazine (RDX) was theoretically investigated at the MP2/cc-PVDZ level. An O-to-N transnitration mechanism was put forward for the N-nitration of N-acetyl tertiary amines, including the formation of diverse complexes R'N(COCH3)RNO2(+) and deacetylate. The electron transfer results in the complex formation, and the acetyl-to-nitro electrophilic displacement leads to deacetylate. Presumably, the carbonyl groups (C═O) in N-acetyl tertiary amines serve as the hinged joint in the electron transfer. Three successive N-nitrations transform TRAT into RDX; their electron transfers are strongly exothermic by -21.1, -19.5, and -15.4 kcal/mol relative to TRAT + 3NO2(+), repectively, and their electrophilic displacements possess low activation Gibbs free energies of 9.0, 6.8, and 7.5 kcal/mol relative to the σ-complexes 6, 11, and 14, respectively. The rate constants of the single electron transfer (SET) and the acetyl-to-nitro displacement were estimated roughly by Marcus and transition-state (TS) theories, respectively, indicating that they are both fast with the strong exothermicity. The available experimental phenomena were well interpreted by the computational results. read less
NOT USED (low confidence) J.-Q. Li, F. Wang, X. Cheng, and X. Li, “Reactive Molecular Dynamics Simulation on Thermal Decomposition of n‐Heptane,” Chinese Journal of Chemical Physics. 2013. link Times cited: 6
Abstract: The thermal decomposition of n‐heptane is an important proce… read more
Abstract: The thermal decomposition of n‐heptane is an important process in petroleum industry. The theoretical investigations show that the main products are C2H4, H2, and C3H6, which agree well with the experimental results. The products populations depend strongly on the temperature. The quantity of ethylene increases quickly as the temperature goes up. The conversion of n‐heptane and the mole fraction of primary products from reactive molecular dynamic and chemical kinetic modeling are compared with each other. We also investigated the pre‐exponential factor and activation energy for thermal decomposition of n‐heptane by kinetic analysis from the reactive force field simulations, which were extracted to be 1.78×1014s−1 and 47.32 kcal/mol respectively. read less
NOT USED (low confidence) J. Ding, L. Zhang, Y. Zhang, and K. Han, “A reactive molecular dynamics study of n-heptane pyrolysis at high temperature.,” The journal of physical chemistry. A. 2013. link Times cited: 104
Abstract: n-Heptane is the most important straight chain paraffin in t… read more
Abstract: n-Heptane is the most important straight chain paraffin in the fossil-fuel industry. In this work, pyrolysis of n-heptane at high temperature is investigated by a series of ReaxFF based reactive molecular dynamic simulations. The pyrolysis correlated intermediate reactions, important product/intermediate distributions, and corresponding kinetics behaviors are systematically analyzed at atomistic level. The results indicate that the entire pyrolysis process is radical-dominated. The unimolecular dissociation is the main pathway of n-heptane decomposition. Initiation of the decomposition is mainly through C-C bond fission. Central C-C bonds would dissociate prior to the terminal ones. Besides, the Rice-Kossiakoff theory is proved for the pyrolysis of n-heptane at the atomistic level. To give a better description of the pyrolysis behavior, some alkane related intermolecular reactions should be considered in the mechanism. The apparent activation energy extracted from the present simulations is 43.02-54.49 kcal/mol in the temperature range 2400-3000 K, which is reasonably consistent with the experimental results. read less
NOT USED (low confidence) K. Elkhodary, S. Tang, and W. K. Liu, “Inclusion clusters in the archetype-blending continuum theory.” 2013. link Times cited: 13
Abstract: In this chapter, we will present a contemporary review of th… read more
Abstract: In this chapter, we will present a contemporary review of the hitherto numerical characterization of nanowires (NWs). The bulk of the research reported in the literatures concern metallic NWs including Al, Cu, Au, Ag, Ni, and their alloys NWs. Research has also been reported for the investigation of some nonmetallic NWs, such as ZnO, GaN, SiC, SiO2. A plenty of researches have been conducted regarding the numerical investigation of NWs. Issues analyzed include structural changes under different loading situations, the formation and propagation of dislocations, and the effect of the magnitude of applied loading on deformation mechanics. Efforts have also been made to correlate simulation results with experimental measurements. However, direct comparisons are difficult since most simulations are carried out under conditions of extremely high strain/loading rates and small simulation samples due to computational limitations. Despite of the immense numerical studies of NWs, a significant work still lies ahead in terms of problem formulation, interpretation of results, identification and delineation of deformation mechanisms, and constitutive characterization of behavior. In this chapter, we present an introduction of the commonly adopted experimental and numerical approaches in studies of the deformation of NWs in Section 1. An overview of findings concerning perfect NWs under different loading situations, such as tension, compression, torsion, and bending are presented in Section 2. In Section 3, we will detail some recent results from the authors’ own work with an emphasis on the study of influences from different pre-existing defect on NWs. Some thoughts on future directions of the computational mechanics of NWs together with Conclusions will be given in the last section. read less
NOT USED (low confidence) Y. Fu and A. To, “Application of Many‐Realization Molecular Dynamics Method to Understand the Physics of Nonequilibrium Processes in Solids.” 2013. link Times cited: 0
NOT USED (low confidence) K. Irikura, “Aminoxyl (nitroxyl) radicals in the early decomposition of the nitramine RDX.,” The journal of physical chemistry. A. 2013. link Times cited: 22
Abstract: The explosive nitramine RDX (1,3,5-trinitrohexahydro-s-triaz… read more
Abstract: The explosive nitramine RDX (1,3,5-trinitrohexahydro-s-triazine) is thought to decompose largely by homolytic N-N bond cleavage, among other possible initiation reactions. Density-functional theory (DFT) calculations indicate that the resulting secondary aminyl (R2N·) radical can abstract an oxygen atom from NO2 or from a neighboring nitramine molecule, producing an aminoxyl (R2NO·) radical. Persistent aminoxyl radicals have been detected in electron-spin resonance (ESR) experiments and are consistent with autocatalytic "red oils" reported in the experimental literature. When the O-atom donor is a nitramine, a nitrosamine is formed along with the aminoxyl radical. Reactions of aminoxyl radicals can lead readily to the "oxy-s-triazine" product (as the s-triazine N-oxide) observed mass-spectrometrically by Behrens and co-workers. In addition to forming aminoxyl radicals, the initial aminyl radical can catalyze loss of HONO from RDX. read less
NOT USED (low confidence) Y. Liu, S. Zybin, J.-S. Guo, A. V. van Duin, and W. Goddard, “Reactive dynamics study of hypergolic bipropellants: monomethylhydrazine and dinitrogen tetroxide.,” The journal of physical chemistry. B. 2012. link Times cited: 18
Abstract: To gain an atomistic-level understanding on physical and che… read more
Abstract: To gain an atomistic-level understanding on physical and chemical processes occurring at the interfaces of hypergolic propellants, we carried out the first reactive dynamic (ReaxFF) simulations to study the reactive hypergolic mixture of monomethylhydrazine (MMH) and dinitrogen tetroxide (NTO), in comparison with the ethanol (EtOH) and NTO mixture that is reactive but nonhypergolic. Our studies show that the MMH-NTO mixture releases energy more rapidly than the EtOH-NTO mixture upon mixing the fuels and oxidizers. We found that the major early chemical reactions between MMH and NTO are hydrogen abstractions and N-N bond scissions. The MMH-NTO mixture has more reaction channels than EtOH-NTO based on statistical analyses of chemical reaction events and channels at early stages of the dynamics. Analyzing the evolution of product distribution over reaction time shows that the oxidizer (NO(2)) diffuses into the fuels (MMH or EtOH) for the occurrence of reactions, demonstrating the influence of physical mixing on chemical reactions. Our simulations suggest that effective hypergolic systems require fuels with low energy barriers of H abstractions and/or bond scissions and oxidizers with large diffusion mobility for efficient physical mixing. read less
NOT USED (low confidence) W. Yan-qing and H. Feng-lei, “Thermal mechanical anisotropic constitutive model and numerical simulations for shocked β-HMX single crystals,” European Journal of Mechanics A-solids. 2012. link Times cited: 16
NOT USED (low confidence) B. D. Jensen, A. Bandyopadhyay, K. Wise, and G. Odegard, “Parametric Study of ReaxFF Simulation Parameters for Molecular Dynamics Modeling of Reactive Carbon Gases.,” Journal of chemical theory and computation. 2012. link Times cited: 35
Abstract: The development of innovative carbon-based materials can be … read more
Abstract: The development of innovative carbon-based materials can be greatly facilitated by molecular modeling techniques. Although the Reax Force Field (ReaxFF) can be used to simulate the chemical behavior of carbon-based systems, the simulation settings required for accurate predictions have not been fully explored. Using the ReaxFF, molecular dynamics (MD) simulations are used to simulate the chemical behavior of pure carbon and hydrocarbon reactive gases that are involved in the formation of carbon structures such as graphite, buckyballs, amorphous carbon, and carbon nanotubes. It is determined that the maximum simulation time step that can be used in MD simulations with the ReaxFF is dependent on the simulated temperature and selected parameter set, as are the predicted reaction rates. It is also determined that different carbon-based reactive gases react at different rates, and that the predicted equilibrium structures are generally the same for the different ReaxFF parameter sets, except in the case of the predicted formation of large graphitic structures with the Chenoweth parameter set under specific conditions. read less
NOT USED (low confidence) M. Warrier, P. Pahari, and S. Chaturvedi, “Interatomic potential parameters for molecular dynamics simulations of RDX using a reactive force field: A validation study,” Journal of Physics: Conference Series. 2012. link Times cited: 2
Abstract: The parameter sets of the ReaxFF potential distributed with … read more
Abstract: The parameter sets of the ReaxFF potential distributed with the open source, Large-scale Atomic/Molecular Massively Parallel Simulator (LAMMPS) code, is validated for simulating crystal RDX. These parameters are used to model crystal RDX and obtain its unit cell size and bulk modulus. It is seen that the parameters supplied with LAMMPS (5-April, 2011 release) do not reproduce the unit cell size and bulk modulus of crystal RDX as reported by experiments and by other simulations using the ReaxFF potential. The simulation method and relevant parts of the LAMMPS code implementing the method has been earlier validated for Cu. We conclude that either the parameter sets provided with the LAMMPS distribution or its implementation of the ReaxFF potential are not suitable for modeling crystal RDX. read less
NOT USED (low confidence) R. W. Conner and D. Dlott, “Time-resolved spectroscopy of initiation and ignition of flash-heated nanoparticle energetic materials,” Journal of Physical Chemistry C. 2012. link Times cited: 23
Abstract: Nanotechnology has brought a great deal of excitement to res… read more
Abstract: Nanotechnology has brought a great deal of excitement to research in energetic materials (EMs). Nanoparticle EMs have high densities of stored energy and the potential for multifunctionality. Here we discuss research on fundamental mechanisms of initation and ignition of EM with Al or B fuel nanoparticles and TeflonAF or nitrocellulose (NC) oxidizer. Polybutadiene (PB) was also used as an inert. The thin-film samples were confined between two windows and were activated by flash-heating the metal nanoparticles with picosecond laser pulses. Reactions of isolated nanoparticles with their surroundings were studied by measuring ablation thresholds. A shock-induced polymer dissociation model was needed to explain the growth of the reaction volume surrounding a flash-heated fuel particle. Thicker oxide passivation layers confined the nanoparticle allowing the pressure to build up to higher values during flash-heating. Initiation, as the onset of chemical reactivity, was probed using time-resolved Raman or infrar... read less
NOT USED (low confidence) A. Jaramillo-Botero, J. Tahir-Kheli, P. V. Allmen, and W. Goddard, “Multiscale, multiparadigm modeling for nano systems characterization and design.” 2012. link Times cited: 1
Abstract: This chapter outlines our progress toward developing a first… read more
Abstract: This chapter outlines our progress toward developing a first-principles-based hierarchical multiscale, multiparadigm modeling and simulation framework for the characterization and optimization of electronic and chemical properties of nanoscale materials and devices. In our approach, we build from the bottom-up by solving the quantum-mechanical (QM) Schrodinger equation for small systems. The results of these calculations lead to physical parameters that feed into methods capable of spanning longer length and time scale with minimum loss of accuracy. This is achieved by having higher-scale quantities self-consistently derived and optimized from the results at finer scales.
In contrast to other methods, we are strictly first-principles-based, and all of our parameters at all scales relate to physically measurable or QM-computable observables. Our approach that is applicable to the forward (materials phenomenology) and inverse (“materials by design”) problems. The inverse problem involves top-down predictions of structures and compositions at a lower scale from desired properties at a higher scale.
The advantages of our strategy over experimental- and phenomenological-based modeling and simulation approaches include the following: (1) providing access to details that are difficult or impossible to measure (e.g., excited electronic states in materials undergoing extreme conditions of pressure, temperature, etc.); (2) the ability to make useful predictions outside the range of experiments (i.e., since all calculations are ultimately related to first principles); and (3) providing sound, first-principles-based, steering for experiments. read less
NOT USED (low confidence) Y. Shin et al., “Variable charge many-body interatomic potentials,” MRS Bulletin. 2012. link Times cited: 56
Abstract: Recent developments in reactive potentials for the simulatio… read more
Abstract: Recent developments in reactive potentials for the simulation of complex bonding and complex chemistry are reviewed. In particular, the reactive force field and charged optimized many-body methods are two paradigms that enable atoms to autonomously determine their charge state and the nature of their local bonding environments. The capabilities of these methods are illustrated by examples involving ionic-covalent systems, a metal-covalent system, a high- k dielectric gate stack, and the interaction of water with an oxide. Prospects for future development and applications are also discussed. read less
NOT USED (low confidence) D. Mathieu, “Formation Enthalpies Derived from Pairwise Interactions: A Step toward More Transferable Reactive Potentials for Organic Compounds.,” Journal of chemical theory and computation. 2012. link Times cited: 4
Abstract: A new approach to the development and parametrization of rea… read more
Abstract: A new approach to the development and parametrization of reactive potentials for organic compounds is put forward. As a byproduct of preliminary efforts in this direction, the performance of a simple representation of the energy of equilibrium structures in term of pairwise atom-atom and bond-bond contributions is investigated. For now, each contribution is assumed constant, given the multiplicity of covalent bonds, rather than computed on-the-fly from geometries and bond orders. In spite of this rough approximation, the approach performs remarkably well by comparison with semiempirical quantum chemical methods. Nevertheless, further refinement proves necessary for some unstable species involved in chemical reactions. As it stands, the present model appears as a promising basis in view of less empirical and more versatile alternatives to group contribution methods for the fast prediction of heats of formation, although much work remains to be done to demonstrate its value as a starting point toward better reactive potentials. read less
NOT USED (low confidence) R. Eason and T. Sewell, “Shock-Induced Inelastic Deformation in Oriented Crystalline Pentaerythritol Tetranitrate,” Journal of Physical Chemistry C. 2012. link Times cited: 32
Abstract: Molecular dynamics simulations were used to study the mechan… read more
Abstract: Molecular dynamics simulations were used to study the mechanisms of shock-induced inelastic deformation in oriented single crystals of the energetic material pentaerythritol tetranitrate (PETN). Supported planar shock waves with Rankine–Hugoniot shock pressures PR–H ∼ 9 GPa were propagated along two different crystal directions: one that is sensitive to initiation ([001]) and another that is relatively insensitive to initiation ([100]). Qualitatively, it was observed that for the sensitive orientation only elastic compression occurred, leading to the propagation of a single wave through the material, whereas for the insensitive direction elastic compression at and immediately behind the shock front was followed by inelastic deformation, leading to a two-wave structure in which the sharp elastic front moves through the crystal at a higher speed than the broader plastic wave. The detailed responses were characterized by calculating several structural and thermal properties including: relative center-of-mass... read less
NOT USED (low confidence) E. Reed, “Electron-Ion Coupling in Shocked Energetic Materials,” Journal of Physical Chemistry C. 2012. link Times cited: 37
Abstract: The magnitude and role of electronic excitations in shocked … read more
Abstract: The magnitude and role of electronic excitations in shocked energetic materials are studied theoretically using quantum molecular dynamics simulations. Focusing on the detonating primary explosive HN3 (hydrazoic acid), this work finds that the material transiently exhibits a high level of electronic excitation characterized by carrier densities in excess of 1021 cm–3, or one electronic excitation for every eight molecules. Electronic excitations enhance the kinetics of chemical decomposition by ∼30%. The electronic heat capacity has a minor impact on the temperatures exhibited, on the order of 100 K. These simulations are performed using the self-consistent charge density functional tight-binding method (SCC-DFTB) combined with a new modification of a multiscale computational scheme for simulation of the coupling between electrons and ions in shocked matter. read less
NOT USED (low confidence) J. Maillet, E. Bourasseau, N. Desbiens, G. Vallverdu, and G. Stoltz, “Mesoscopic simulations of shock-to-detonation transition in reactive liquid high explosive,” EPL (Europhysics Letters). 2011. link Times cited: 35
Abstract: An extension of the model described in a previous work (see … read more
Abstract: An extension of the model described in a previous work (see Maillet J. B. et al., EPL, 78 (2007) 68001) based on Dissipative Particle Dynamics is presented and applied to a liquid high explosive (HE), with thermodynamic properties mimicking those of liquid nitromethane. Large scale nonequilibrium simulations of reacting liquid HE with model kinetic under sustained shock conditions allow a better understanding of the shock-to-detonation transition in homogeneous explosives. Moreover, the propagation of the reactive wave appears discontinuous since ignition points in the shocked material can be activated by the compressive waves emitted from the onset of chemical reactions. read less
NOT USED (low confidence) A. P. Garcia, D. Sen, and M. Buehler, “Nature’s Flexible and Tough Armor.” 2011. link Times cited: 1
NOT USED (low confidence) X.-F. Chen, B.-Z. Wang, and K. Han, “A reaction of formaldehyde with acetonitrile: understanding the preparation of RDX (I),” RSC Advances. 2011. link Times cited: 3
Abstract: The reaction of HCHO with CH3CN to yield a key RDX precursor… read more
Abstract: The reaction of HCHO with CH3CN to yield a key RDX precursor (TRAT) was proposed to undergo four sequential stages. More attention was suggested to be paid to the mid and final terms. A parallel product could be controlled by using the concentrated sulfuric acid in almost drying conditions. read less
NOT USED (low confidence) K. D. Smith, M. Bruns, S. Stoliarov, M. Nyden, O. Ezekoye, and P. R. Westmoreland, “Assessing the effect of molecular weight on the kinetics of backbone scission reactions in polyethylene using Reactive Molecular Dynamics,” Polymer. 2011. link Times cited: 23
NOT USED (low confidence) Y. Fu, M. Kırca, and A. To, “On determining the thermal state of individual atoms in molecular dynamics simulations of nonequilibrium processes in solids,” Chemical Physics Letters. 2011. link Times cited: 6
NOT USED (low confidence) Q.-D. Wang, J. Wang, J.-Q. Li, N. Tan, and X. Li, “Reactive molecular dynamics simulation and chemical kinetic modeling of pyrolysis and combustion of n-dodecane,” Combustion and Flame. 2011. link Times cited: 173
NOT USED (low confidence) M. Warrier, P. Pahari, and S. Chaturvedi, “Validation Of A Reactive Force Field Included With An Open Source, Massively Parallel Code For Molecular Dynamics Simulations Of RDX.” 2010. link Times cited: 0
Abstract: Molecular dynamics (MD) simulations of RDX is carried out us… read more
Abstract: Molecular dynamics (MD) simulations of RDX is carried out using the ReaxFF force field supplied with the Large‐scale Atomic/Molecular Massively Parallel Simulator (LAMMPS). Validation of ReaxFF to model RDX is carried out by extracting the (i) crystal unit cell parameters, (ii) bulk modulus and (iii) thermal expansion coefficient and comparing with reported values from both experiments and simulations. read less
NOT USED (low confidence) W. Goddard, J. Mueller, K. Chenoweth, and A. Duin, “ReaxFF Monte Carlo reactive dynamics. Application to resolving the partial occupations of the M1 phase of the MoVNbTeO catalyst,” Catalysis Today. 2010. link Times cited: 27
NOT USED (low confidence) L. Zhang, S. Zybin, A. V. van Duin, and W. Goddard, “Modeling High Rate Impact Sensitivity of Perfect RDX and HMX Crystals by ReaxFF Reactive Dynamics,” Journal of Energetic Materials. 2010. link Times cited: 42
Abstract: We report a methodology for rapid assessment of impact sensi… read more
Abstract: We report a methodology for rapid assessment of impact sensitivity of energetic materials which uses the ReaxFF reactive force field in reactive dynamics (RD) simulations of the high rate compression/expansion of a perfect energetic crystal. This approach is validated here to study the high rate impact sensitivity of 1,3,5-trinitrohexahydro-s-triazine (RDX) crystal and octahydro-1,3,5,7-tetrazocine (HMX) crystal at different phases (α, β, γ, and δ). These simulations found that for a compression rate of ∼8.76 km/s along the [100] direction, RDX crystals at lower volume compression ratios (x = 30, 35, 38%) led only to a few RDX molecules decomposing and only into primary products. However, at higher compression ratios (x ≥ 40%), all RDX molecules in the crystal decompose very quickly, leading to both primary and secondary decomposition reactions, including various intermediates such as NO2, NO, HONO, and OH and final products such as H2O, N2, CO, and CO2. For the various phases of HMX, these ReaxFF RD simulations found noticeably higher impact sensitivity for the δ-phase than for other three phases (α, β, and γ). At the same compression ratio x = 40%, all HMX molecules in δ-phase decompose leading to both primary and secondary reaction. However, at 40%, only few HMX molecules in α-, β-, and γ-phases decompose. For a higher compression ratio (x = 42%), increased HMX decomposition is observed for all four phases. These simulation results for both RDX and HMX crystals agree qualitatively with experiment observations. We also observe a variation of the strain energy in different HMX phases induced by the high rate compression, which could be related to the sensitivity difference of HMX phases. These simulations typically took less than 18 h to run on a single 3.0-GHz processor, demonstrating that the fast compression approach by MD simulations with the ReaxFF force field can be used for a quick evaluation of the sensitivity of energetic materials. read less
NOT USED (low confidence) S. Zybin, P. Xu, Q. An, and W. G. III, “ReaxFF Reactive Molecular Dynamics: Coupling Mechanical Impact to Chemical Initiation in Energetic Materials,” 2010 DoD High Performance Computing Modernization Program Users Group Conference. 2010. link Times cited: 4
Abstract: We report an approach to large-scale atomistic simulations o… read more
Abstract: We report an approach to large-scale atomistic simulations of chemical initiation processes in shocked energetic materials based on a parallel implementation of the ReaxFF reactive force field. Here we present results of Compressive Shear Reactive Dynamics (CSRD) simulations on compressed PETN and RDX single crystal, conventional high explosives. We show that CSRD can evaluate anisotropy of shock sensitivity along different crystallographic directions in single crystal explosives and provide estimates in agreement with experiment for PETN and RDX. The temperature increase is much faster for shear along the slip planes related to experimentally sensitive shock directions. We also investigate the effect of shear on chemical initiation. The dominant initiation reactions in both systems is NO2 dissociation whose rate significantly varies along shock and slip directions. All calculations are performed with the massively parallel MD code GRASP enabling multi-million atom reactive MD simulations of chemical processes in many important stockpile materials. read less
NOT USED (low confidence) E. Reed, “Atomic transformation pathways from terahertz radiation generated by shock-induced phase transformations,” Physical Review B. 2010. link Times cited: 4
Abstract: This work shows that detectable terahertz (THz) frequency ra… read more
Abstract: This work shows that detectable terahertz (THz) frequency radiation can be emitted when a wurtzite-structure crystal transforms to a rocksalt structure under shock compression on picosecond time scales. Information about the atomic-scale transformation pathway is contained in the sign of the emitted THz electric field and information about the kinetics is contained in the time dependence. This phenomenon provides an avenue to experimental measurement of microscopic transformation pathways in crystals on the shortest (picosecond) time scales. read less
NOT USED (low confidence) F. Guo, H. Zhang, and X. Cheng, “MOLECULAR DYNAMIC SIMULATIONS OF SOLID NITROMETHANE UNDER HIGH PRESSURES,” Journal of Theoretical and Computational Chemistry. 2010. link Times cited: 18
Abstract: We report ReaxFF molecular dynamic simulations of structure … read more
Abstract: We report ReaxFF molecular dynamic simulations of structure change of crystalline nitromethane and the formation of hydrogen bond under high pressure. Under high pressure, the angles between C–N bonds and X, Y and Z axes have changed. Through the calculation of g(r) of O and H atoms, we found a new peak near 1.6 A, which indicates the formation of the hydrogen bond between O and H atoms. We calculated the distribution of the angles of the C–N bonds orientations, the distribution of the dihedral angle of CNOO, and the charge distribution of nitromethane molecules under various pressures, and made a comparison between low and high pressures. The effects of hydrogen bonding in high explosive materials are discussed. read less
NOT USED (low confidence) N. Goldman, E. Reed, and L. Fried, “Quantum mechanical corrections to simulated shock Hugoniot temperatures.,” The Journal of chemical physics. 2009. link Times cited: 41
Abstract: We present a straightforward method for the inclusion of qua… read more
Abstract: We present a straightforward method for the inclusion of quantum nuclear vibrational effects in molecular dynamics calculations of shock Hugoniot temperatures. Using a Gruneisen equation of state and a quasiharmonic approximation to the vibrational energies, we derive a simple, postprocessing method for calculation of the quantum corrected Hugoniot temperatures. We have used our novel technique on ab initio simulations of shock compressed water and methane. Our results indicate significantly closer agreement with all available experimental temperature data for these two systems. Our formalism can be easily applied to a number of different shock compressed molecular liquids or solids, and has the potential to decrease the large uncertainties inherent in many experimental Hugoniot temperature measurements of these systems. read less
NOT USED (low confidence) D. Bedrov, J. Hooper, G. D. Smith, and T. Sewell, “Shock-induced transformations in crystalline RDX: a uniaxial constant-stress Hugoniostat molecular dynamics simulation study.,” The Journal of chemical physics. 2009. link Times cited: 49
Abstract: Molecular dynamics (MD) simulations of uniaxial shock compre… read more
Abstract: Molecular dynamics (MD) simulations of uniaxial shock compression along the [100] and [001] directions in the alpha polymorph of hexahydro-1,3,5-trinitro-1,3,5-triazine (alpha-RDX) have been conducted over a wide range of shock pressures using the uniaxial constant stress Hugoniostat method [Ravelo et al., Phys. Rev. B 70, 014103 (2004)]. We demonstrate that the Hugoniostat method is suitable for studying shock compression in atomic-scale models of energetic materials without the necessity to consider the extremely large simulation cells required for an explicit shock wave simulation. Specifically, direct comparison of results obtained using the Hugoniostat approach to those reported by Thompson and co-workers [Phys. Rev. B 78, 014107 (2008)] based on large-scale MD simulations of shocks using the shock front absorbing boundary condition (SFABC) approach indicates that Hugoniostat simulations of systems containing several thousand molecules reproduced the salient features observed in the SFABC simulations involving roughly a quarter-million molecules, namely, nucleation and growth of nanoscale shear bands for shocks propagating along the [100] direction and the polymorphic alpha-gamma phase transition for shocks directed along the [001] direction. The Hugoniostat simulations yielded predictions of the Hugoniot elastic limit for the [100] shock direction consistent with SFABC simulation results. read less
NOT USED (low confidence) S. S. Han, J. L. Mendoza-Cortes, and W. A. Goddard, “Recent advances on simulation and theory of hydrogen storage in metal-organic frameworks and covalent organic frameworks.,” Chemical Society reviews. 2009. link Times cited: 419
Abstract: This critical review covers the application of computer simu… read more
Abstract: This critical review covers the application of computer simulations, including quantum calculations (ab initio and DFT), grand canonical Monte-Carlo simulations, and molecular dynamics simulations, to the burgeoning area of the hydrogen storage by metal-organic frameworks and covalent-organic frameworks. This review begins with an overview of the theoretical methods obtained from previous studies. Then strategies for the improvement of hydrogen storage in the porous materials are discussed in detail. The strategies include appropriate pore size, impregnation, catenation, open metal sites in metal oxide parts and within organic linker parts, doping of alkali elements onto organic linkers, substitution of metal oxide with lighter metals, functionalized organic linkers, and hydrogen spillover (186 references). read less
NOT USED (low confidence) E. Salmon, A. Duin, F. Lorant, P. Marquaire, and W. Goddard, “Thermal decomposition process in algaenan of Botryococcus braunii race L. Part 2: Molecular dynamics simulations using the ReaxFF reactive force field,” Organic Geochemistry. 2009. link Times cited: 95
NOT USED (low confidence) A. Page and B. Moghtaderi, “Molecular dynamics simulation of the low-temperature partial oxidation of CH4.,” The journal of physical chemistry. A. 2009. link Times cited: 44
Abstract: Low-temperature partial oxidation of methane was investigate… read more
Abstract: Low-temperature partial oxidation of methane was investigated using reactive molecular dynamics (MD) and quantum mechanical (QM) methods. In particular, the ReaxFF hydrocarbon force field [Chenoweth, K.; et al. J. Phys. Chem. A 2008, 112, 1040] was employed to simulate a [20 CH(4) + 10 O(2)] model system at 500 degrees C. The chemical mechanism of the partial oxidation of methane was proposed on the basis of analysis of the computed trajectory of this model system. The partial oxidation of methane was observed to be initiated by the abstraction of hydrogen from CH(4) by O(2) and the atomization of CH(4) itself. Subsequent radical recombination between hydrogen atoms and the dehydrogenation of CH(4) were the primary pathways by which H(2) was formed. In agreement with current models of low-temperature combustion, radicals including H(3)C-OO and H(2)C-OO were also observed during the MD simulation. The observed reaction mechanism was subsequently analyzed using QM methods. For instance, structural features of prominent radical species observed during the MD simulation were analyzed using density functional theory (DFT) and coupled-cluster (CCSD(T)) methods. Enthalpies of reaction of all observed chemical processes were calculated using DFT and the W1 composite method. Where possible, comparisons with experimental data were made. read less
NOT USED (low confidence) Y.-C. Chen, K. Nomura, R. Kalia, A. Nakano, and P. Vashishta, “Molecular dynamics nanoindentation simulation of an energetic material,” Applied Physics Letters. 2008. link Times cited: 17
Abstract: Molecular dynamics simulation approach is used to study nano… read more
Abstract: Molecular dynamics simulation approach is used to study nanoindentation of the (100) crystal surface of cyclotrimethylenetrintramine (RDX) by a diamond indenter. The indenter and substrate atoms interact via reactive force fields. Nanoindentation causes significant heating of the RDX substrate in the proximity of the indenter, resulting in the release of molecular fragments and subsequent “walking” motion of these molecules on the indenter surfaces. read less
NOT USED (low confidence) P. J. Donoghue, P. Helquist, P. Norrby, and O. Wiest, “Development of a Q2MM Force Field for the Asymmetric Rhodium Catalyzed Hydrogenation of Enamides.,” Journal of chemical theory and computation. 2008. link Times cited: 61
Abstract: The rhodium catalyzed asymmetric hydrogenation of enamides t… read more
Abstract: The rhodium catalyzed asymmetric hydrogenation of enamides to generate amino acid products and derivatives is a widely used method to generate unnatural amino acids. The choice of a chiral ligand is of utmost importance in this reaction and is often based on high throughput screening or simply trial and error. A virtual screening method can greatly increase the speed of the ligand screening process by calculating expected enantiomeric excesses from relative energies of diastereomeric transition states. Utilizing the Q2MM method, new molecular mechanics parameters are derived to model the hydride transfer transition state in the reaction. The new parameters were based off of structures calculated at the B3LYP/LACVP** level of theory and added to the MM3* force field. The new parameters were validated against a test set of experimental data utilizing a wide range of bis-phosphine ligands. The computational model agreed with experimental data well overall, with an unsigned mean error of 0.6 kcal/mol against a set of 18 data points from experiment. The major errors in the computational model were due either to large energetic errors at high e.e., still resulting in qualitative agreement, or cases where large steric interactions prevent the reaction from proceeding as expected. read less
NOT USED (low confidence) K. Chenoweth, A. Duin, P. Persson, M. Cheng, J. Oxgaard, and W. Goddard, “Development and application of a ReaxFF reactive force field for oxidative dehydrogenation on vanadium oxide catalysts (The Journal of Physical Chemistry A (2008) 112C),” Journal of Physical Chemistry A. 2008. link Times cited: 127
Abstract: We have developed a new ReaxFF reactive force field to descr… read more
Abstract: We have developed a new ReaxFF reactive force field to describe accurately reactions of hydrocarbons with vanadium oxide catalysts. The ReaxFF force field parameters have been fit to a large quantum mechanics (QM) training set containing over 700 structures and energetics related to bond dissociations, angle and dihedral distortions, and reactions between hydrocarbons and vanadium oxide clusters. In addition, the training set contains charge distributions for small vanadium oxide clusters and the stabilities of condensed-phase systems. We find that ReaxFF reproduces accurately the QM training set for structures and energetics of small clusters. Most important is that ReaxFF describes accurately the energetics for various oxidation states of the condensed phases, including V2O5, VO2, and V2O3 in addition to metallic V (V0). To demonstrate the capability of the ReaxFF force field for describing catalytic processes involving vanadium oxides, we performed molecular dynamics (MD) simulation for reactions of a ... read less
NOT USED (low confidence) J. Patterson, Z. Dreger, M. Miao, and Y. Gupta, “Shock wave induced decomposition of RDX: time-resolved spectroscopy.,” The journal of physical chemistry. A. 2008. link Times cited: 58
Abstract: Time-resolved optical spectroscopy was used to examine chemi… read more
Abstract: Time-resolved optical spectroscopy was used to examine chemical decomposition of RDX crystals shocked along the [111] orientation to peak stresses between 7 and 20 GPa. Shock-induced emission, produced by decomposition intermediates, was observed over a broad spectral range from 350 to 850 nm. A threshold in the emission response of RDX was found at about 10 GPa peak stress. Below this threshold, the emission spectrum remained unchanged during shock compression. Above 10 GPa, the emission spectrum changed with a long wavelength component dominating the spectrum. The long wavelength emission is attributed to the formation of NO2 radicals. Above the 10 GPa threshold, the spectrally integrated intensity increased significantly, suggesting the acceleration of chemical decomposition. This acceleration is attributed to bimolecular reactions between unreacted RDX and free radicals. These results provide a significant experimental foundation for further development of a decomposition mechanism for shocked RDX (following paper in this issue). read less
NOT USED (low confidence) J. Leininger, C. Minot, and F. Lorant, “Two theoretical simulations of hydrocarbons thermal cracking: Reactive force field and density functional calculations,” Journal of Molecular Structure-theochem. 2008. link Times cited: 18
NOT USED (low confidence) S. Zhao, T. Germann, and A. Strachan, “Molecular Dynamics Characterization of the Response of Ni/Al Nanolaminates Under Dynamic Loading,” Journal of Propulsion and Power. 2007. link Times cited: 8
Abstract: We use a recently developed molecular dynamics method with a… read more
Abstract: We use a recently developed molecular dynamics method with an accurate, first-principles-based force field to study shock propagation in Ni/Al nanolaminates and the induced (highly exothermic) chemical reactions. We characterize both perfect nanolaminates and specimens containing small (4-nm diameter) voids. The new method enables the accurate description of both the nonequilibrium shock-loading process and the long time evolution of the shocked material, providing an atomic-level picture of the complex interplay between the mechanical, thermal, and chemical processes that govern the behavior of the metastable composites. We shock the nanolaminates in the direction normal to the Ni/Al interfaces, leading to multiple wave reflections, due to the elastic mismatch between Ni and Al; this leads to the Al layers having a higher temperature during the early stages of the process. In the perfect nanolaminates, the chemical reactions start at the interfaces closest to the impact plane and then propagate through the material. A rapid increase in the rate of chemical reactions (3Ni + Al → Ni 3 Al) is observed following the melting of the Ni and Al layers. We estimate the propagation velocity of the chemical front to be about 200 m/s. The porous samples exhibit much faster energy-release rates, due to the mechanical intermixing of Al and Ni caused by shock-induced pore collapse and the higher shock temperatures. read less
NOT USED (low confidence) E. Reed et al., “Terahertz radiation from shocked materials,” Materials Today. 2007. link Times cited: 10
NOT USED (low confidence) J. Maillet, L. Soulard, and G. Stoltz, “A reduced model for shock and detonation waves. II. The reactive case,” EPL (Europhysics Letters). 2007. link Times cited: 31
Abstract: We present a mesoscopic model for reactive shock waves, whic… read more
Abstract: We present a mesoscopic model for reactive shock waves, which extends the model proposed in G. Stoltz, Europhys. Lett., 76 (2006) 849. A complex molecule (or a group of molecules) is replaced by a single mesoparticle, evolving according to some Dissipative Particle Dynamics. Chemical reactions can be handled in a mean way by considering an additional variable per particle describing the progress of the reaction. The evolution of the progress variable is governed by the kinetics of a reversible exothermic reaction. Numerical results give profiles in qualitative agreement with all-atom studies. read less
NOT USED (low confidence) S. Stoliarov, R. Lyon, and M. Nyden, “A reactive molecular dynamics model of thermal decomposition in polymers. II. Polyisobutylene,” Polymer. 2004. link Times cited: 51
NOT USED (low confidence) J. Dick, D. Hooks, R. Menikoff, and A. R. Martinez, “Elastic-plastic wave profiles in cyclotetramethylene tetranitramine crystals,” Journal of Applied Physics. 2004. link Times cited: 109
Abstract: The explosive molecular crystal cyclotetramethylene tetranit… read more
Abstract: The explosive molecular crystal cyclotetramethylene tetranitramine was studied in three orientations in a set of plate impact experiments; the orientations studied were {110}, {011}, and {010} in P21/n space group. The elastic–plastic shock response was measured using laser interferometry. The measured particle velocity profiles showed elastic precursor decay typical of a stress relaxing material. There is anisotropy in elastic shock strength and decay. The amount of precursor decay with propagation distance and stress relaxation behind the elastic shock varied among the orientations. The {010} orientation had larger elastic precursors than did the other two orientations; the {010} crystal does not have the regular plastic deformation mechanisms available to it. Elastic Hugoniots were obtained from the measurements. The inelastic deformation mechanisms may vary with orientation. read less
NOT USED (low confidence) D. Hong, Y. Guo, C. Wang, and R. Wei, “Coal/NH3 interactions during co-pyrolysis and their effects on the char reactivity for NO-reduction: A ReaxFF MD study,” Fuel. 2023. link Times cited: 5
NOT USED (low confidence) M. Zheng, X. Li, J. Bai, and L. Guo, “Chemical structure effects on coal pyrolyzates and reactions by using large-scale reactive molecular dynamics,” Fuel. 2022. link Times cited: 9
NOT USED (low confidence) C. M. Ashraf, S. Shabnam, Y. Xuan, and A. Duin, “Application of ReaxFF-Reactive Molecular Dynamics and Continuum Methods in High-Temperature/Pressure Pyrolysis of Fuel Mixtures,” Computational Approaches for Chemistry Under Extreme Conditions. 2019. link Times cited: 2
NOT USED (low confidence) X. Chen and C. Goldsmith, “Predictive kinetics for the thermal decomposition of RDX,” Proceedings of the Combustion Institute. 2019. link Times cited: 37
NOT USED (low confidence) M. Islam, M. Cherukara, E. Antillon, and A. Strachan, “Shock-Induced Chemistry: Molecular Dynamics and Coarse Grain Modeling,” Computational Approaches for Chemistry Under Extreme Conditions. 2019. link Times cited: 5
NOT USED (low confidence) E. Martínez, E. Kober, and M. Cawkwell, “Accelerated Molecular Dynamics Simulations of Shock-Induced Chemistry: Application to Liquid Benzene,” Computational Approaches for Chemistry Under Extreme Conditions. 2019. link Times cited: 0
NOT USED (low confidence) S. Winczewski, M. Y. Shaheen, and J. Rybicki, “Interatomic potential suitable for the modeling of penta-graphene: Molecular statics/molecular dynamics studies,” Carbon. 2018. link Times cited: 34
NOT USED (low confidence) S. McGrane, P. Bowlan, K. E. Brown, C. Bolme, and M. Cawkwell, “Broadband mid-infrared measurements for shock induced chemistry,” Bulletin of the American Physical Society. 2018. link Times cited: 5
NOT USED (low confidence) M. Radue, “Molecular Modeling of Aerospace Polymer Matrices Including Carbon Nanotube-Enhanced Epoxy.” 2017. link Times cited: 2
NOT USED (low confidence) D. Hong, H. Shu, X. Guo, and C. Zheng, “Molecular Dynamics Simulations Study of Brown Coal Pyrolysis Using ReaxFF Method.” 2016. link Times cited: 4
NOT USED (low confidence) L. K. Harper, “Computational Investigation of Pernicious Compounds: Arsenic and High Energy Density Materials and Their relevant Mechanisms.” 2015. link Times cited: 0
Abstract: COMPUTATIONAL INVESTIGATION OF PERNICIOUS COMPOUNDS: ARSENIC… read more
Abstract: COMPUTATIONAL INVESTIGATION OF PERNICIOUS COMPOUNDS: ARSENIC AND HIGH ENERGY DENSITY MATERIALS AND THEIR RELEVANT MECHANISMS Lenora Kathleen Harper Old Dominion University, 2015 Director: Dr. Craig A. Bayse The underlying mechanisms of chemical warfare agents (CWAs) and high energy density materials (HEDMs) are poorly understood, yet important to the development of chemical applications in military science and technology. The reactivity of arsenite and arsenic-based CWA lewisite w ith biological thiols plays an important role in toxicity of these elements. Toxic effects of arsenite are eliminated when arsenite and selenite are co-administered, due to their antagonistic relationship. The arsenic-based CWA lewisite is detoxified by the dithiol-containing British anti-lewisite (BAL). The reduction of arsenous acid by thiol, the formation of an As-Se species, and the detoxification of lewisite w ith BAL have been modeled using density functional theory (DFT) and solventassisted proton exchange (SAPE), a microsolvation technique that uses a network of water molecules to mimic the participation of bulk solvent in proton transfer processes. Several pathways were explored for the formation o f an As-Se bond with the nucleophilic attack o f selenide on (RS)2AsOH to form (RS^AsSeH as the most likely, consistent with previous experimental studies. DFT-SAPE activation barriers for the twostep detoxification of lewisite with BAL predict rapid formation of a ring product, also observed experimentally. The initial step in the decomposition of HEDMs is not yet understood due to the rapid rate at which the reaction takes place. Trigger bonds, or bonds that break to initiate explosive decomposition, were characterized using Wiberg bond indices (WBIs) for known HEDMs RDX, HMX, TNT, PETN, for comparison to recently-synthesized tetrazole-based explosives. WBIs were compared to reference compounds to determine the degree of weakening in bond strength (AWBI) to show that N-N02, C-N02, and 0N02 bonds are likely to cleave initially, consistent w ith previous experimental and theoretical results. Tetrazole-based molecules w ith small side chains and one -N 0 2 group are predicted to have an N-N02 trigger bond. Larger side chains w ith one -N 0 2 group are predicted to break in the C-N side chain backbone. Molecules w ith more than one —N02 will cleave at the C-N bond from tetrazole. This dissertation is dedicated to my parents, Leonard and Tamrae Harper read less
NOT USED (low confidence) L. Huang and J. Kieffer, “Challenges in modeling mixed ionic-covalent glass formers.” 2015. link Times cited: 11
NOT USED (low confidence) M. Manaa and L. Fried, “The Reactivity of Energetic Materials Under High Pressure and Temperature,” Advances in Quantum Chemistry. 2014. link Times cited: 19
NOT USED (low confidence) D. Taylor and B. Rice, “Quantum-Informed Multiscale M&S for Energetic Materials,” Advances in Quantum Chemistry. 2014. link Times cited: 10
NOT USED (low confidence) C. M. Berg et al., “Experiments probing fundamental mechanisms of energetic material initiation and ignition,” MRS Proceedings. 2012. link Times cited: 0
NOT USED (low confidence) P. Kennedy, B. Garrison, M. F. Russo, and A. V. van Duin, “Strategies for modeling diverse chemical reactions in molecular dynamics simulations of cluster bombardment,” Surface and Interface Analysis. 2011. link Times cited: 1
Abstract: Reaction energies for the degradation reactions of poly(meth… read more
NOT USED (low confidence) M. Buehler, “Computational Scale Linking in Biological Protein Materials.” 2010. link Times cited: 0
NOT USED (low confidence) S. Keten, J. Bertaud, D. Sen, Z. Xu, T. Ackbarow, and M. Buehler, “Multiscale Modeling of Biological Protein Materials – Deformation and Failure.” 2010. link Times cited: 1
NOT USED (low confidence) S. Mishra and M. Meuwly, “Reactive Processes with Molecular Simulations.” 2010. link Times cited: 2
NOT USED (low confidence) B. Rice and T. Sewell, “Equilibrium Molecular Dynamics Simulations.” 2009. link Times cited: 12
NOT USED (low confidence) M. Meyers, H. Jarmakani, E. Bringa, and B. Remington, “Chapter 89 Dislocations in Shock Compression and Release.” 2009. link Times cited: 54
NOT USED (low confidence) M. Buehler, “Hierarchical Nanomechanics of Collagen Fibrils: Atomistic and Molecular Modeling.” 2008. link Times cited: 26
NOT USED (low confidence) E. Reed, M. Manaa, L. Fried, K. Glaesemann, and J. Joannopoulos, “A transient semimetallic layer in detonating nitromethane,” Nature Physics. 2008. link Times cited: 123
NOT USED (low confidence) D. Mathieu and A. Lucas, “Computational approaches to the dynamics of ions and electrons in materials under extreme conditions,” Computational Materials Science. 2007. link Times cited: 5
NOT USED (low confidence) D. Moore, S. McGrane, and D. J. Funk, “Ultrashort Laser Shock Dynamics.” 2007. link Times cited: 5
NOT USED (low confidence) B. Holian, T. Germann, A. Strachan, and J. Maillet, “Non-Equilibrium Molecular Dynamics Studies of Shock and Detonation Processes in Energetic Materials.” 2005. link Times cited: 2
NOT USED (low confidence) W. Goddard, “A Perspective of Materials Modeling.” 2005. link Times cited: 9
NOT USED (low confidence) D. Moore, D. J. Funk, and S. McGrane, “At the Confluence of Experiment and Simulation: Ultrafast Laser Spectroscopic Studies of Shock Compressed Energetic Materials.” 2005. link Times cited: 3
NOT USED (low confidence) D. Sorescu, S. Alavi, and D. Thompson, “Theoretical and Computational Studies of Energetic Salts.” 2005. link Times cited: 4
NOT USED (high confidence) B. Hamilton, M. Kroonblawd, J. Macatangay, H. Springer, and A. Strachan, “Intergranular Hotspots: A Molecular Dynamics Study on the Influence of Compressive and Shear Work,” The Journal of Physical Chemistry C. 2023. link Times cited: 3
Abstract: Numerous crystal- and microstructural-level mechanisms are a… read more
Abstract: Numerous crystal- and microstructural-level mechanisms are at play in the formation of hotspots, which are known to govern high explosive initiation behavior. Most of these mechanisms, including pore collapse, interfacial friction, and shear banding, involve both compressive and shear work done within the material and have thus far remained difficult to separate. We assess hotspots formed at shocked crystal-crystal interfaces using quasi-1D molecular dynamics simulations that isolate effects due to compression and shear. Two high explosive materials are considered (TATB and PETN) that exhibit distinctly different levels of molecular conformational flexibility and crystal packing anisotropy. Temperature and intra-molecular strain energy localization in the hotspot is assessed through parametric variation of the crystal orientation and two velocity components that respectively modulate compression and shear work. The resulting hotspots are found to be highly localized to a region within 5-20 nm of the crystal-crystal interface. Compressive work plays a considerably larger role in localizing temperature and intra-molecular strain energy for both materials and all crystal orientations considered. Shear induces a moderate increase in energy localization relative to unsheared cases only for relatively weak compressive shock pressures of approximately 10 GPa. These results help isolate and rank the relative importance of hotspot generation mechanisms and are anticipated to guide the treatment of crystal-crystal interfaces in coarse-grained models of polycrystalline high explosive materials. read less
NOT USED (high confidence) M. Zhou, G. Wei, Y. Zhang, D. Xiang, and C. Ye, “Molecular dynamic insight into octahydro-1,3,5,7-tetranitro-1,3,5,7-tetrazocine (HMX) and the nano-HMX decomposition mechanism,” RSC Advances. 2022. link Times cited: 0
Abstract: Herein, we demonstrate the use of large-scale reactive molec… read more
Abstract: Herein, we demonstrate the use of large-scale reactive molecular dynamics simulations to identify the influence of nanostructures, size effects, and temperature for the decomposition processes of octahydro-1,3,5,7-tetranitro-1,3,5,7-tetrazocine (HMX). The bulk-phase and six types of HMX nanoparticle (30–70 Å) systems were investigated at two high temperatures (2000 K and 3000 K). The evolution of the potential energy (PE) and total number of molecules (TM) of HMX crystals and their six nanoparticle systems were analyzed and addressed, and it was revealed that the nanostructure has a great accelerative effect on the thermal decomposition of HMX. The temperature distribution, initial decomposition process, and main intermediate and gas products were traced, and showed that the initial decomposition of HMX nanoparticles is triggered by the dissociation of the N–NO2 bond. With the increase in temperature, the total amount of gas molecules in HMX nanoparticles rapidly increases, which shows that the high temperature can accelerate the decomposition rate for HMX nanoparticles. read less
NOT USED (high confidence) Z. Fthenakis, I. Petsalakis, V. Tozzini, and N. Lathiotakis, “Evaluating the performance of ReaxFF potentials for sp2 carbon systems (graphene, carbon nanotubes, fullerenes) and a new ReaxFF potential,” Frontiers in Chemistry. 2022. link Times cited: 7
Abstract: We study the performance of eleven reactive force fields (Re… read more
Abstract: We study the performance of eleven reactive force fields (ReaxFF), which can be used to study sp2 carbon systems. Among them a new hybrid ReaxFF is proposed combining two others and introducing two different types of C atoms. The advantages of that potential are discussed. We analyze the behavior of ReaxFFs with respect to 1) the structural and mechanical properties of graphene, its response to strain and phonon dispersion relation; 2) the energetics of (n, 0) and (n, n) carbon nanotubes (CNTs), their mechanical properties and response to strain up to fracture; 3) the energetics of the icosahedral C60 fullerene and the 40 C40 fullerene isomers. Seven of them provide not very realistic predictions for graphene, which made us focusing on the remaining, which provide reasonable results for 1) the structure, energy and phonon band structure of graphene, 2) the energetics of CNTs versus their diameter and 3) the energy of C60 and the trend of the energy of the C40 fullerene isomers versus their pentagon adjacencies, in accordance with density functional theory (DFT) calculations and/or experimental data. Moreover, the predicted fracture strain, ultimate tensile strength and strain values of CNTs are inside the range of experimental values, although overestimated with respect to DFT. However, they underestimate the Young’s modulus, overestimate the Poisson’s ratio of both graphene and CNTs and they display anomalous behavior of the stress - strain and Poisson’s ratio - strain curves, whose origin needs further investigation. read less
NOT USED (high confidence) M. Rahm, “Electronegativity at the Shock Front,” Propellants, Explosives, Pyrotechnics. 2022. link Times cited: 0
Abstract: : In this work, a scale for pressure-adapted atomic electron… read more
Abstract: : In this work, a scale for pressure-adapted atomic electronegativity is used to make general predictions of bond polarity in H -, C -, N - and O -based compounds experiencing shock conditions. The qualitative picture that emerges is one of increasing polarity of several bonds common in energetic materials. The general predictions made are compared to, and found to support, claims of ionic decomposition routes in compressed nitromethane and nitrate esters at high pressure. Changing electronegativity is also suggested as a factor driving the ionic dis-proportionation of various molecular phases with compression. Calculations using the eXtreme-Pressure Polar-izable Continuum Model (XP-PCM) predict increasing energy differences between ground and excited states in non-bonded H , C , N , and O atoms as a function of pressure. This data enables for a discussion on the reliability of elec-tronegativity-based rationales at more extreme thermodynamic conditions. read less
NOT USED (high confidence) B. Hamilton, M. Kroonblawd, and A. Strachan, “Extemporaneous Mechanochemistry: Shock-Wave-Induced Ultrafast Chemical Reactions Due to Intramolecular Strain Energy.,” The journal of physical chemistry letters. 2022. link Times cited: 12
Abstract: Regions of energy localization referred to as hotspots are k… read more
Abstract: Regions of energy localization referred to as hotspots are known to govern shock initiation and the run-to-detonation in energetic materials. Mounting computational evidence points to accelerated chemistry in hotspots from large intramolecular strains induced via the interactions between the shock wave and microstructure. However, definite evidence mapping intramolecular strain to accelerated or altered chemical reactions has so far been elusive. From a large-scale reactive molecular dynamics simulation of the energetic material 1,3,5-triamino-2,4,6-trinitrobenzene, we map decomposition kinetics to molecular temperature and intramolecular strain energy prior to reaction. Both temperature and intramolecular strain are shown to accelerate chemical kinetics. A detailed analysis of the atomistic trajectory shows that intramolecular strain can induce a mechanochemical alteration of decomposition mechanisms. The results in this paper could inform continuum-level chemistry models to account for a wide range of mechanochemical effects. read less
NOT USED (high confidence) B. Hamilton, M. Kroonblawd, and A. Strachan, “The Potential Energy Hotspot: Effects of Impact Velocity Defect Geometry, and Crystallographic Orientation.” 2021. link Times cited: 7
Abstract: In energetic materials, the localization of energy into “hot… read more
Abstract: In energetic materials, the localization of energy into “hotspots” when a shock wave interacts with the material’s microstructure is known to dictate the initiation of chemical reactions and detonation. Recent results have shown that, following the shock-induced collapse of pores with circular cross-sections, more energy is localized as internal potential energy (PE) than can be inferred from the kinetic energy (KE) distribution. This leads to a complex thermo-mechanical state that is typically overlooked. The mechanisms associated with pore collapse and hotspot formation and the resulting energy localization are known to be highly dependent on material properties, especially its ability to deform plastically and alleviate strain energy, as well as the size and shape of the pore. Therefore, we use molecular dynamics simulations to characterize shockinduced pore collapse and the subsequent formation of hotspots in TATB, a highly anisotropic molecular crystal, for various defect shapes, shock strengths and crystallographic orientations. We find that the the localization of energy as PE is consistently higher and its extent larger than as localized as KE. A detailed analysis of the MD trajectories reveal the underlying molecular process that govern the effect of orientation and pore shape on the resulting hotspots. We find that the regions of highest PE for a given KE relate to not the impact front of the collapse, but the areas of maximum plastic deformation, while KE is maximized at the point of impact. A comparison with previous results in HMX reveal less energy localization in TATB which could be a contributing factor to its insensitivity. read less
NOT USED (high confidence) J. Hu, Z. Wilde, P. Peralta, C. Muhich, and J. Oswald, “Predicting Hugoniot equation of state in erythritol with ab initio and reactive molecular dynamics,” Journal of Applied Physics. 2021. link Times cited: 0
Abstract: Erythritol has been proposed as an inert surrogate for devel… read more
Abstract: Erythritol has been proposed as an inert surrogate for developing theoretical and computational models to study aging in energetic materials. In this work, we present a comparison of mechanical and shock properties of erythritol computed using the ReaxFF reactive force field and from ab initio calculations employing density functional theory (DFT). We screened eight different ReaxFF parameterizations, of which the CHO parameters developed for hydrocarbon oxidation provide the most accurate predictions of mechanical properties and the crystal structure of erythritol. Further validation of the applicability of this ReaxFF parameterization for modeling erythritol is demonstrated by comparing predictions of the elastic constants, crystal structure, vibrational density of states, and Hugoniot curves against DFT calculations. The ReaxFF predictions are in close agreement with the DFT simulations for the elastic constants and shock Hugoniot when the crystal is loaded along its c axis but show as much as 30% disagreement in the elastic constants in the a b plane and 12% difference in shock pressures when shocked along the a or b crystal axes. Last, we compare thermomechanical properties predicted from classical molecular dynamics with those calculated using the quasi-harmonic approximation and show that quantum mechanical effects produce large discrepancies in the computed values of heat capacity and thermal expansion coefficients compared with classical assumptions. Combining classical molecular dynamics predictions of mechanical behavior with phonon-based calculations of thermal behaviors, we show that predicted shock-induced temperatures for pressures up to 6.5 GPa do not exceed the pressure-dependent melting point of erythritol. read less
NOT USED (high confidence) C. Wang, Y. Ni, C. Zhang, and X. Xue, “Crystal Structure Prediction of 2,4,6,8,10,12-Hexanitro-2,4,6,8,10,12-hexaazaisowurtzitane (CL-20) by a Tailor-Made OPLS-AA Force Field,” Crystal Growth & Design. 2021. link Times cited: 9
Abstract: Predicting the crystal structure of an energetic compound is… read more
Abstract: Predicting the crystal structure of an energetic compound is an important aspect of energetic crystal engineering because an accurate crystal structure prediction (CSP) presents a base for accurate... read less
NOT USED (high confidence) F. Zhang, Y. Cao, X. Liu, H. Xu, D. Lu, and R. Yang, “How Small Molecules Affect the Thermo-Oxidative Aging Mechanism of Polypropylene: A Reactive Molecular Dynamics Study,” Polymers. 2021. link Times cited: 6
Abstract: Understanding the aging mechanism of polypropylene (PP) is f… read more
Abstract: Understanding the aging mechanism of polypropylene (PP) is fundamental for the fabrication and application of PP-based materials. In this paper, we present our study in which we first used reactive molecular dynamics (RMD) simulations to explore the thermo-oxidative aging of PP in the presence of acetic acid or acetone. We studied the effects of temperature and oxygen on the aging process and discussed the formation pathways of typical small molecule products (H2, CO, CO2, CH4, C2H4, and C2H6). The effect of two infection agents, acetic acid and acetone, on the aging reaction was analyzed emphatically. The simulation results showed that acetone has a weak impact on accelerating the aging process, while acetic acid has a significant effect, consistent with previous experimental studies. By tracking the simulation trajectories, both acetic acid and acetone produced small active free radicals to further react with other fragment products, thus accelerating the aging process. The first reaction step of acetic acid is often the shedding of the H atom on the hydroxyl group, while the reaction of acetone is often the shedding of the H atom or the methyl. The latter requires higher energy at lower temperatures. This is why the acceleration effect of acetone for the thermo-oxidative aging of PP was not so significant compared to acetic acid in the experimental temperature (383.15 K). read less
NOT USED (high confidence) B. Li, J. Jiang, L.-L. Li, and J.-F. Peng, “Thermal stability and detonation character of nitroso-substituted derivatives of cubane,” Molecular Physics. 2021. link Times cited: 2
Abstract: ABSTRACT A series of derivatives of nitroso-substituted cuba… read more
Abstract: ABSTRACT A series of derivatives of nitroso-substituted cubane were designed through the substitution of hydrogen atoms by nitroso groups one by one. The heats of formation (HOFs) were calculated to explore the thermal stability. The bond dissociation energies (BDEs) and the bond orders of the trigger bonds were also investigated to explore the molecular stability kinetically. Furthermore, the steric effect was confirmed as the determinant of molecular stability for title molecules. To explore the detonation properties, the detonation pressure (P), the detonation velocity (D), the heat of detonation (Q), and the specific density (ρ) were calculated by using the empirical Kamlet-Jacobs (K-J) equations. To predict the sensitivity, the characteristic drop height (H 50) is calculated. Based on our calculations, the octanitrosocubane is found with detonation parameters (D = 9.03 km/s, P = 36.96 GPa) better than that of RDX with enough stability. GRAPHICAL ABSTRACT read less
NOT USED (high confidence) Y. Xiao, L. Chen, K. Yang, D. Geng, J. Lu, and J. Wu, “Mechanism of the improvement of the energy of host–guest explosives by incorporation of small guest molecules: HNO3 and H2O2 promoted C–N bond cleavage of the ring of ICM-102,” Scientific Reports. 2021. link Times cited: 6
NOT USED (high confidence) L. Komissarov, R. Rüger, M. Hellström, and T. Verstraelen, “ParAMS: Parameter Optimization for Atomistic and Molecular Simulations,” Journal of chemical information and modeling. 2021. link Times cited: 8
Abstract: This work introduces ParAMS-a versatile Python package that … read more
Abstract: This work introduces ParAMS-a versatile Python package that aims to make parametrization workflows in computational chemistry and physics more accessible, transparent, and reproducible. We demonstrate how ParAMS facilitates the parameter optimization for potential energy surface (PES) models, which can otherwise be a tedious specialist task. Because of the package's modular structure, various functionality can be easily combined to implement a diversity of parameter optimization protocols. For example, the choice of PES model and the parameter optimization algorithm can be selected independently. An illustration of ParAMS' strengths is provided in two case studies: (i) a density functional-based tight binding (DFTB) repulsive potential for the inorganic ionic crystal ZnO and (ii) a ReaxFF force field for the simulation of organic disulfides. read less
NOT USED (high confidence) P. Yoo, M. Sakano, S. Desai, M. M. Islam, P. Liao, and A. Strachan, “Neural network reactive force field for C, H, N, and O systems,” npj Computational Materials. 2021. link Times cited: 30
NOT USED (high confidence) F. Wang, L. Chen, D. Geng, J. Lu, and J. Wu, “Chemical reactions of a CL-20 crystal under heat and shock determined by ReaxFF reactive molecular dynamics simulations.,” Physical chemistry chemical physics : PCCP. 2020. link Times cited: 10
Abstract: Studying the chemical reactions of hexanitrohexaazaisowurtzi… read more
Abstract: Studying the chemical reactions of hexanitrohexaazaisowurtzitane (CL-20) under heat and shock is helpful to understand its sensitivity and shock initiation mechanism. In this work, several molecular dynamics simulations were performed under three different conditions: high temperature, high temperature and pressure, and shock. The formation and breakage of chemical bonds, changes of bond lengths, and initial reactions were analysed. It was found that the main small-molecule product of CL-20 during initial decomposition under the three different conditions was always NO2, but the generation pathways were different. At high temperatures, NO2 was generated by the direct cleavage of N-NO2 bonds. In contrast, high pressure and shock promoted the transfer of O atoms to N atoms connected to NO2, leading to the breakage of N-NO2 bonds. Almost all NO2 originated from the transfer of O atoms under the shock conditions. read less
NOT USED (high confidence) M. M. Islam and A. Strachan, “Role of dynamical compressive and shear loading on hotspot criticality in RDX via reactive molecular dynamics,” Journal of Applied Physics. 2020. link Times cited: 16
Abstract: Energy localization in hotspots due to shock-induced pore co… read more
Abstract: Energy localization in hotspots due to shock-induced pore collapse is thought to be a critical process in the initiation of heterogeneous high-energy density materials. The dynamical collapse of porosity involves expansion, jetting, shearing, and recompression of the material surrounding the defect. While the resulting hotspots are known to result in deflagration waves that can lead to detonation, we lack the understanding of the relative potency of the various processes that occur during the collapse. We use molecular dynamics simulations with the reactive force field ReaxFF to characterize how uniaxial expansion/recompression, shear, and combinations thereof affect the formation and criticality of hotspots in RDX, 1,3,5-trinitro-1,3,5-triazine. We chose a planar pore configuration consisting of a 40 nm gap and independently control the relative amounts of compressive and shear shock loadings. We find that shear-dominated critical hotspots tend to be smaller but exhibit higher temperatures than uniaxial ones and involve longer reaction time scales. Interestingly, the chemical decomposition mechanisms are affected by the relative amount of dynamical shear and uniaxial loads. read less
NOT USED (high confidence) M. Izadi, A. Maghari, W. Zhang, and A. V. van Duin, “Reactive molecular dynamics simulation for isotope-exchange reactions in H/D systems: ReaxFFHD development.,” The Journal of chemical physics. 2020. link Times cited: 1
Abstract: To investigate the chemical isotope-exchange reactions withi… read more
Abstract: To investigate the chemical isotope-exchange reactions within a system composed of a mixture of hydrogen and deuterium (H/D) in the plasma media, the ReaxFFHD potential was parameterized against an appropriate quantum mechanics (QM)-based training set. These QM data involve structures and energies related to bond dissociation, angle distortion, and an exchange reaction of the tri-atomic molecular ions, H3 +, D3 +, H2D+, and D2H+, produced in the hydrogen plasma. Using the ReaxFFHD potential, a range of reactive molecular dynamics simulations were performed on different mixtures of H/D systems. Analysis of the reactions involved in the production of these tri-atomic molecular ions was carried out over 1 ns simulations. The results show that the ReaxFFHD potential can properly model isotope-exchange reactions of tri-atomic molecular ions and that it also has a perfect transferability to reactions taking place in these systems. In our simulations, we observed some intermediate molecules (H2, D2, and HD) that undergo secondary reactions to form the tri-atomic molecular ions as the most likely products in the hydrogen plasma. Moreover, there remains a preference for D in the produced molecular ions, which is related to the lower zero-point energy of the D-enriched species, showing the isotope effects at the heart of the ReaxFFHD potential. read less
NOT USED (high confidence) A. Islam, M. S. Islam, N. Ferdous, J. Park, and A. Hashimoto, “Vacancy-induced thermal transport in two-dimensional silicon carbide: a reverse non-equilibrium molecular dynamics study.,” Physical chemistry chemical physics : PCCP. 2020. link Times cited: 23
Abstract: Because of its impressive electrical, thermal, and mechanica… read more
Abstract: Because of its impressive electrical, thermal, and mechanical properties, two-dimensional silicon carbide (2D-SiC) has recently gained tremendous attention in the field of nanoelectronics and optoelectronics. Here, we investigated the effects of various types of defects such as bi-, point-, and mixed-vacancies on the thermal conductivity of 2D-SiC using reverse non-equilibrium molecular dynamics simulation. The effects of temperature variation on the thermal conductivity of vacancy-defected 2D-SiC were also studied. A significant reduction of the thermal conductivity was observed when the concentrations of the vacancies were increased. The point vacancy resulted in the thermal conductivity decreasing more quickly as compared to bi vacancy and mixed vacancy defects. Moreover, increasing the temperature of vacancy-defected 2D-SiC further reduced the thermal conductivity due to a strong phonon-vacancy scattering effect. Because of the introduction of vacancy defects in the acoustic phonon density of states (PDOS), a softening behavior in the intensity of the characteristic peaks is perceived, and with increasing temperature, a frequency shrinking is noted in the PDOS curves, both of which contribute to the reduction of the thermal conductivity. Additionally, rapid softening of the phonon transmission spectrum and increase in entropy were obtained for the point vacancy-defected structure, which clearly confirms our findings at different vacancy concentrations as well as for types of vacancies. These findings are very much imperative for realizing heat dissipation in nano- and optoelectronic devices based on 2D-SiC as well as for demonstrating an effective method for modulating 2D-SiC thermal conductivity through defect engineering. read less
NOT USED (high confidence) L. W. Bertels, L. B. Newcomb, M. Alaghemandi, J. R. Green, and M. Head‐Gordon, “Benchmarking the Performance of the ReaxFF Reactive Force Field on Hydrogen Combustion Systems.,” The journal of physical chemistry. A. 2020. link Times cited: 24
Abstract: A thorough understanding of the kinetics and dynamics of com… read more
Abstract: A thorough understanding of the kinetics and dynamics of combusting mixtures is of considerable interest, especially in regimes beyond the reach of current experimental validation. The ReaxFF reactive force field method has provided a way to simulate large-scale systems of hydrogen combustion via a parameterized potential that can simulate bond breaking. This modeling approach has been applied to hydrogen combustion, as well as myriad other reactive chemical systems. In this work, we benchmark the performance of several common parameterizations of this potential against higher-level quantum mechanical (QM) approaches. We demonstrate instances where these parameterizations of the ReaxFF potential fail both quantitatively and qualitatively to describe reactive events relevant for hydrogen combustion systems. read less
NOT USED (high confidence) L. Jiang et al., “Study of the thermal decomposition mechanism of FOX-7 by molecular dynamics simulation and online photoionization mass spectrometry,” RSC Advances. 2020. link Times cited: 7
Abstract: The thermal decomposition mechanism of energetic materials i… read more
Abstract: The thermal decomposition mechanism of energetic materials is important for analyzing the combustion mechanisms of propellants and evaluating the safety of propellants during transport and storage. 1,1-Diamino-2,2-dinitroethylene (FOX-7) is an important insensitive energetic material that can be used as an oxidizer in propellants. However, the initial decomposition mechanism of FOX-7 is not clear to date. The ReaxFF molecular dynamics method is widely used in the investigation of the thermal decomposition mechanisms of energetic materials. Meanwhile, the combination of thermogravimetry with online photoionization time-of-flight mass spectrometry (TG-PI-TOF-MS) and online single-photon ionization time-of-flight mass spectrometry (SPI-TOF-MS) can reveal the decomposition products, which may be integrated with the results of the simulation. In this study, the primary thermal decomposition mechanism of 1,1-diamino-2,2-dinitroethylene (FOX-7) was studied by the ReaxFF molecular dynamics simulations and online photoionization mass spectrometry. The results of the molecular dynamics simulations showed that the primary decomposition step of FOX-7 is C–NO2 cleavage; after this, C
O formation occurs via a three-membered ring transition state, followed by NO elimination. The remaining structure loses NH2 and H, resulting in the formation of the NHCCO structure, which finally breaks down into HNC and CO. NH2 reacts with an H atom to produce NH3. A reversible intramolecular hydrogen transfer was also observed at 2500 K; however, it failed to dominate the decomposition reaction. During the decomposition of FOX-7, the major products are N2, NH3, CO2, and H2N2 and the minor products are H2O, HN2, and H2. The TG-PI-TOF-MS spectrum shows three signals, i.e., m/z = 18, 28, and 30, which can be assigned to H2O, CO, and NO, respectively. Moreover, four signals at m/z = 72.72, 55.81, 45.79, and 29.88 corresponding to the products (NH2)2CCO, (NH2)CCO, NO2, and NO have been obtained in the SPI-TOF-MS spectrum. The experimental data obtained via online photoionization mass spectrometry further validated the results of the molecular dynamics simulations. read less
NOT USED (high confidence) C. Tang et al., “Another transfer channel for shock energy flowing into intra-molecular: the coherent transfer channel,” Physica Scripta. 2020. link Times cited: 1
Abstract: In this paper, we have proposed that there is another transf… read more
Abstract: In this paper, we have proposed that there is another transfer channel for shock energy flowing into to intra-molecular vibration mode. During this energy transfer process, these higher frequency vibrations would gradually possess a coherence at the direction of shock wave. Namely, these optical phonons could be coherent after shock wave compressed. Based on the probe light can be scattered by the coherent optical phonons, a series of light scattering experiments at varying detection angles were performed to explore the existing evidence of coherent optical phonon in shocked materials. No matter the incident angle is 6.2° or 15.7°, the Signal intensity ratio γs and the line width Γ of compressed vibration were both showed peak value around the corresponding phase matching angle θ ′ , which was attributed to the contribution of coherent optical phonons. Taken together, these experimental results demonstrated the existence of new proposed shock energy transfer channel, that coherent transfer channel, and open new perspectives in the molecular dissociation channel research in shocked materials. read less
NOT USED (high confidence) S.-H. Zhu et al., “First-principles study of structural, elastic, electronic and optical properties of RDX under pressure,” Philosophical Magazine. 2020. link Times cited: 1
Abstract: ABSTRACT The influences of pressure on structural, elastic, … read more
Abstract: ABSTRACT The influences of pressure on structural, elastic, electronic and optical properties of α-RDX under pressure from 0 to 40 GPa have been investigated by performing first-principles calculations. The obtained structural parameters based on the GGA-PBE+G calculations are consistent with previous experimental values. The results of B/G, C12-C44 and Poisson's ratio show that α-RDX has changed to ductility under pressure between 0 and 5 GPa. The obvious rotation of NO2 group in the equatorial position appears, especially in the range of pressure from 10 to 15 GPa, which influences the elastic and mechanical properties of α-RDX. Moreover, we find that the electrons of α-RDX become more active under higher pressure by comparing the curves of DOS under different pressure. Furthermore, the anisotropy of optical properties under different pressures has been shown. read less
NOT USED (high confidence) R. Benda, G. Zucchi, É. Cancès, and B. Lebental, “Insights into the π - π interaction driven non-covalent functionalization of carbon nanotubes of various diameters by conjugated fluorene and carbazole copolymers.,” The Journal of chemical physics. 2020. link Times cited: 12
Abstract: We investigate the interaction of polyfluorene and fluorene/… read more
Abstract: We investigate the interaction of polyfluorene and fluorene/carbazole copolymers bearing various functional groups and side chains with small to large diameter-from 1.7 nm to 9 nm-carbon nanotubes (CNTs) in vacuo. We use variable-charge molecular dynamics simulations based on the reactive force field ReaxFF. We show that non-covalent functionalization of nanotubes, driven by π - π interactions, is effective for all the polymers studied, thanks to their conjugated backbone and regardless of the presence of specific functional groups. The geometry at equilibrium of these polymer/CNT hybrids is analyzed in detail at the scale of each fluorene or carbazole unit. The role of both the functional groups and the alkyl chain length is analyzed in detail. Adsorption of the polymers on the nanotube sidewalls is shown to be either complete-with the whole chain physisorbed-or partial-due to intrachain coiling or interchain repulsion-depending on the initial geometry, number of polymers, and nanotube diameter. Energetic arguments supplement the described geometric features. Both energetic and geometric adsorption features are derived here for the first time for large diameter carbon nanotubes (up to 9 nm) and fluorene/carbazole copolymers having up to 30 monomers and bearing different functional groups. The force field ReaxFF and its available parameterization used for the simulations are validated, thanks to a benchmark and review on higher-level quantum calculations-for simple π - π interacting compounds made up of polycyclic aromatic molecules adsorbed on a graphene sheet or bilayer graphene. Although it is shown that the influence of the nanotube chirality on the adsorption pattern and binding strength cannot be discussed with our method, we highlight that an available force field such as ReaxFF and its parameterization can be transferable to simulate new systems without specific re-parameterization, provided that this model is validated against reference methods or data. This methodology proves to be a valuable tool for optimal polymer design for nanotube functionalization at no re-parameterization cost and could be adapted to simulate and assist the design of other types of molecular systems. read less
NOT USED (high confidence) C. Ren, H. Liu, X. Li, and L. Guo, “Decomposition mechanism scenarios of CL-20 co-crystals revealed by ReaxFF molecular dynamics: similarities and differences.,” Physical chemistry chemical physics : PCCP. 2020. link Times cited: 18
Abstract: Understanding the similarities and differences of decomposit… read more
Abstract: Understanding the similarities and differences of decomposition mechanisms of CL-20 and its cocrystals is of great interest for practical applications of CL-20 cocrystals. The responses of CL-20 cocrystals to thermal stimulus were investigated using ReaxFF molecular dynamics simulations of two representative cocrystals, CL-20/HMX and CL-20/TNT, under adiabatic conditions and comparing to the baseline system of pure CL-20. The comprehensive chemical details were revealed with the aid of the unique code of VARxMD. The three CL-20-involved reactive systems all exhibit a distinct three-stage character during adiabatic decomposition when using the double peaks of the major intermediate NO2 amount as the boundary. By taking advantage of the three-stage classification, a clear scenario for the similar stimulus-response of the CL-20 cocrystals can be elucidated for the dominant primary decomposition of CL-20 in stage I and the transition of favored chemical mechanisms from the generation of intermediates/radicals in stage II into their consumption to form stable products in stage III. The similar chemical behaviors are rooted in the dominance of CL-20 chemistry in the initial response of its cocrystals to thermal stimulus. The prolonged reaction zone uncovers the slowed decomposition kinetics of CL-20/HMX and CL-20/TNT, which is associated with the altered kinetics of CL-20 decomposition specifically by N-NO2 bond scission and CL-20 skeleton decay. The retarded CL-20 decomposition in its cocrystals consequently results in more moderate self-heating and less violent exothermic reactions that agrees with the experimental observations of improved stability and damaged detonation performance of CL-20 cocrystals, particularly for CL-20/TNT. The results obtained in this work suggest that ReaxFF MD simulations can provide useful insight for the modulated chemical properties of varied CL-20 cocrystals. read less
NOT USED (high confidence) X.-yan Wang, X. Zhang, Y. Song, Z.-lin Xu, Y. Meng, and B. Li, “Theoretical exploration about nitro-substituted derivatives of pyrimidine as high-energy-density materials,” Journal of Molecular Modeling. 2019. link Times cited: 4
NOT USED (high confidence) F. Miao and X. Cheng, “Effect of electric field on polarization and decomposition of RDX molecular crystals: a ReaxFF molecular dynamics study,” Journal of Molecular Modeling. 2019. link Times cited: 19
NOT USED (high confidence) J. Zeng, L. Cao, C.-H. Chin, H. Ren, J. Z. Zhang, and T. Zhu, “ReacNetGenerator: an automatic reaction network generator for reactive molecular dynamics simulations.,” Physical chemistry chemical physics : PCCP. 2019. link Times cited: 8
Abstract: Reactive molecular dynamics (MD) simulation makes it possibl… read more
Abstract: Reactive molecular dynamics (MD) simulation makes it possible to study the reaction mechanism of complex reaction systems at the atomic level. However, the analysis of MD trajectories which contain thousands of species and reaction pathways has become a major obstacle to the application of reactive MD simulation in large-scale systems. Here, we report the development and application of the Reaction Network Generator (ReacNetGenerator) method. It can automatically extract the reaction network from the reaction trajectory without any predefined reaction coordinates and elementary reaction steps. Molecular species can be automatically identified from the cartesian coordinates of atoms and the hidden Markov model is used to filter the trajectory noises which makes the analysis process easier and more accurate. The ReacNetGenerator has been successfully used to analyze the reactive MD trajectories of the combustion of methane and 4-component surrogate fuel for rocket propellant 3 (RP-3), and it has great advantages in terms of efficiency and accuracy compared to traditional manual analysis. read less
NOT USED (high confidence) A. Islam et al., “Anomalous temperature dependent thermal conductivity of two-dimensional silicon carbide,” Nanotechnology. 2019. link Times cited: 46
Abstract: Recently, two-dimensional silicon carbide (2D-SiC) has attra… read more
Abstract: Recently, two-dimensional silicon carbide (2D-SiC) has attracted considerable interest due to its exotic electronic and optical properties. Here, we explore the thermal properties of 2D-SiC using reverse non-equilibrium molecular dynamics simulation. At room temperature, a thermal conductivity of ∼313 W mK−1 is obtained for 2D-SiC which is one order higher than that of silicene. Above room temperature, the thermal conductivity deviates the normal 1/T law and shows an anomalous slowly decreasing behavior. To elucidate the variation of thermal conductivity, the phonon modes at different length and temperature are quantified using Fourier transform of the velocity auto-correlation of atoms. The calculated phonon density of states at high temperature shows a shrinking and softening of the peaks, which induces the anomaly in the thermal conductivity. On the other hand, quantum corrections are applied to avoid the freezing effects of phonon modes on the thermal conductivity at low temperature. In addition, the effect of potential on the thermal conductivity calculation is also studied by employing original and optimized Tersoff potentials. These findings provide a means for better understating as well as designing the efficient thermal management of 2D-SiC based electronics and optoelectronics in near future. read less
NOT USED (high confidence) K. Zheng et al., “The solid phase thermal decomposition and nanocrystal effect of hexahydro-1,3,5-trinitro-1,3,5-triazine (RDX) via ReaxFF large-scale molecular dynamics simulation.,” Physical chemistry chemical physics : PCCP. 2019. link Times cited: 13
Abstract: The solid phase thermal decomposition and nanocrystal effect… read more
Abstract: The solid phase thermal decomposition and nanocrystal effect are extremely important to understand the ignition, combustion, reaction growth and buildup to detonation under shock wave action. To explore the basic mechanism at the atomic level and understand the interaction among nanocrystal lattices, molecules, and intermediates during the solid phase decomposition, ReaxFF large-scale molecular dynamics simulation at 1000-3000 K was demonstrated on the solid phase of nanocrystalline RDX with a size in the range of 5-12 nm. Based on the analysis of the RDX decay and chemical species, we found that the whole decomposition process can be divided into the solid-affected stage and the following less-condensed phase stage. From the results of NO2 diffusion and high frequency reaction statistics for the nanocrystal effect on the RDX decay, intermediate diffusion was found to be strongly associated with the chemical pathway. In addition, it was found for the first time that the thermal decomposition of RDX originates from the inside of the nanocrystal instead of its surface. Furthermore, a promising uniform energy distribution mechanism transfer by vibration inside the nanocrystalline RDX was demonstrated. The detailed information derived from this study can aid in the thorough understanding of the size effect on the chemical kinetics of nanoexplosives, especially for thermal decomposition and reaction growth. read less
NOT USED (high confidence) E. Martinez et al., “Parallel replica dynamics simulations of reactions in shock compressed liquid benzene.,” The Journal of chemical physics. 2019. link Times cited: 9
Abstract: The study of the long-term evolution of slow chemical reacti… read more
Abstract: The study of the long-term evolution of slow chemical reactions is challenging because quantum-based reactive molecular dynamics simulation times are typically limited to hundreds of picoseconds. Here, the extended Lagrangian Born-Oppenheimer molecular dynamics formalism is used in conjunction with parallel replica dynamics to obtain an accurate tool to describe the long-term chemical dynamics of shock-compressed benzene. Langevin dynamics has been employed at different temperatures to calculate the first reaction times in liquid benzene at pressures and temperatures consistent with its unreacted Hugoniot. Our coupled engine runs for times on the order of nanoseconds (one to two orders of magnitude longer than traditional techniques) and is capable of detecting reactions that are characterized by rates significantly slower than we could study before. At lower pressures and temperatures, we mainly observe Diels-Alder metastable reactions, whereas at higher pressures and temperatures we observe stable polymerization reactions. read less
NOT USED (high confidence) L. Yue, L. Lv, Z. Xu, L. Zhang, and M. Yang, “A reactive force field molecular dynamics study of molecular nitrogen and water mixtures under high temperature and high pressure,” Journal of Molecular Modeling. 2019. link Times cited: 2
NOT USED (high confidence) Y. Long and J. Chen, “Theoretical study of the microscopic Doppler effect for energetic material,” Philosophical Magazine. 2019. link Times cited: 0
Abstract: ABSTRACT We develop a physical model to describe the microsc… read more
Abstract: ABSTRACT We develop a physical model to describe the microscopic Doppler effect of phonon states in energetic material and use it to investigate the phonon–strain scattering behaviour of β-octahydro-1,3,5,7-tetranitro-1,3,5,7-tetrazocine. The required elastic constants and force constants are obtained by first-principles calculations. By using the phonon–strain scattering probability, a set of dissipation parameters are calculated, such as the viscosity coefficient, damping rate of elastic wave, and heat dissipation across shock wave front. It is interesting that the Doppler effect could describe the microscopic phonon scattering mechanism reasonably. read less
NOT USED (high confidence) W. Zhang, D. Dong, D. Bedrov, and A. V. van Duin, “Hydroxide transport and chemical degradation in anion exchange membranes: a combined reactive and non-reactive molecular simulation study,” Journal of Materials Chemistry A. 2019. link Times cited: 22
Abstract: Investigating the structural and dynamical properties, charg… read more
Abstract: Investigating the structural and dynamical properties, charge transport and membrane degradation in anion exchange membranes (AEMs) using atomistic-scale simulations provides a guideline for the design of new high-performance membrane fuel cells. read less
NOT USED (high confidence) Y. Long and J. Chen, “Theoretical study of the defect evolution for molecular crystal under shock loading,” Journal of Applied Physics. 2019. link Times cited: 13
Abstract: We simulate the shock loading process of β-octahydro-1,3,5,7… read more
Abstract: We simulate the shock loading process of β-octahydro-1,3,5,7-tetranitro-1,3,5,7-tetrazocine by molecular dynamics and calculate the isoentropic curve, Hugoniot curve, temperature field, velocity field, stress field, and density field. Based on the simulation results, we develop a physical model to describe the pore collapse, crack expansion, and hot spot formation mechanisms and calculate a set of key parameters, such as pore collapsing speed, rarefaction wave speed, and crack expansion speed. A microscopic physical picture for defect evolution at the early time of shock loading is obtained. read less
NOT USED (high confidence) D. Ilyin, W. A. Goddard, J. J. Oppenheim, and T. Cheng, “First-principles–based reaction kinetics from reactive molecular dynamics simulations: Application to hydrogen peroxide decomposition,” Proceedings of the National Academy of Sciences. 2018. link Times cited: 29
Abstract: This paper presents our vision of how to use in silico appro… read more
Abstract: This paper presents our vision of how to use in silico approaches to extract the reaction mechanisms and kinetic parameters for complex condensed-phase chemical processes that underlie important technologies ranging from combustion to chemical vapor deposition. The goal is to provide an analytic description of the detailed evolution of a complex chemical system from reactants through various intermediates to products, so that one could optimize the efficiency of the reactive processes to produce the desired products and avoid unwanted side products. We could start with quantum mechanics (QM) to ensure an accurate description; however, to obtain useful kinetics we need to average over ∼10-nm spatial scales for ∼1 ns, which is prohibitively impractical with QM. Instead, we use the reactive force field (ReaxFF) trained to fit QM to carry out the reactive molecular dynamics (RMD). We focus here on showing that it is practical to extract from such RMD the reaction mechanisms and kinetics information needed to describe the reactions analytically. This analytic description can then be used to incorporate the correct reaction chemistry from the QM/ReaxFF atomistic description into larger-scale simulations of ∼10 nm to micrometers to millimeters to meters using analytic approaches of computational fluid dynamics and/or continuum chemical dynamics. In the paper we lay out the strategy to extract the mechanisms and rate parameters automatically without the necessity of knowing any details of the chemistry. We consider this to be a proof of concept. We refer to the process as RMD2Kin (reactive molecular dynamics to kinetics) for the general approach and as ReaxMD2Kin (ReaxFF molecular dynamics to kinetics) for QM-ReaxFF–based reaction kinetics. read less
NOT USED (high confidence) M. Meuwly, “Reactive molecular dynamics: From small molecules to proteins,” Wiley Interdisciplinary Reviews: Computational Molecular Science. 2018. link Times cited: 33
Abstract: The current status of reactive molecular dynamics (MD) simul… read more
Abstract: The current status of reactive molecular dynamics (MD) simulations is summarized. Both, methodological aspects and applications to problems ranging from gas phase reaction dynamics to ligand‐binding in solvated proteins are discussed, focusing on extracting information from simulations that cannot easily be obtained from experiments alone. One specific example is the structural interpretation of the ligand rebinding time scales extracted from state‐of‐the art time‐resolved experiments. Atomistic simulations employing validated reactive interaction potentials are capable of providing structural information about the time scales involved. Both, merits and shortcomings of the various methods are discussed and the outlook summarizes possible future avenues such as reactive potentials based on machine learning techniques. read less
NOT USED (high confidence) F. Yin, C. Tang, Q. Wang, X. Liu, and Y. Tang, “Molecular Dynamics Simulations on the Thermal Decomposition of Meta-Aramid Fibers,” Polymers. 2018. link Times cited: 19
Abstract: The thermal decomposition mechanism of a meta-aramid fiber w… read more
Abstract: The thermal decomposition mechanism of a meta-aramid fiber was simulated at the atomic level using the ReaxFF reactive force field. The simulation results indicated that the main initial decomposition positions of the meta-aramid fiber elements were Caromatic ring–N and C=O, which could be used as targets for the modification of meta-aramid fibers. The meta-aramid fiber elements first decomposed into C6–C13 and then into smaller segments and micromolecular gases. The temperature was shown to be the key factor affecting the thermal decomposition of the meta-aramid fibers. More complex compositions and stable gases were produced at high temperatures than at lower temperatures. HCN was a decomposition product at high temperature, suggesting that its presence could be used for detecting thermal faults in meta-aramid fibers. Generation path tracing of the thermal decomposition products NH3 and H2O was also performed. NH3 was produced when the NH2 group captured an H atom adjacent to the system. H2O was formed after a carbonyl group captured an H atom, became a hydroxyl group, with subsequent intramolecular dehydration or intermolecular hydrogen abstraction. read less
NOT USED (high confidence) N. Ge, S. Bai, J. Chang, and G. Ji, “Shock response of condensed-phase RDX: molecular dynamics simulations in conjunction with the MSST method,” RSC Advances. 2018. link Times cited: 6
Abstract: We have performed molecular dynamics simulations in conjunct… read more
Abstract: We have performed molecular dynamics simulations in conjunction with the multiscale shock technique (MSST) to study the initial chemical processes of condensed-phase RDX under various shock velocities (8 km s−1, 10 km s−1 and 11 km s−1). A self-consistent charge density functional tight-binding (SCC-DFTB) method was used. We find that the N–NO2 bond dissociation is the primary pathway for RDX with the NO2 groups facing (group 1) the shock, whereas the C–N bond scission is the dominant primary channel for RDX with the NO2 groups facing away from (group 2) the shock. In addition, our results present that the NO2 groups facing away from the shock are rather inert to shock loading. Moreover, the reaction pathways of a single RDX molecule under the 11 km s−1 shock velocity have been mapped out in detail, NO2, NO, N2O, CO and N2 were the main products. read less
NOT USED (high confidence) K. Song, G. Ji, K. M. Kumari, and D. Wei, “Blending effect between n-decane and toluene in oxidation: a ReaxFF study,” Molecular Simulation. 2018. link Times cited: 3
Abstract: We studied dependency of toluene oxidation-blended n-decane … read more
Abstract: We studied dependency of toluene oxidation-blended n-decane on blending ratio and temperature using the reactive molecular dynamics (RMD) simulations with the newly developed reactive force field (ReaxFF). Different initial reaction pathways of toluene were observed between pure and blended toluene, while that of n-decane showed little contrast. The differences in toluene oxidation paths are related to radical pool, which is largely influenced by H/C ratio. We analysed the influence of H/C ratio on the consumption of intermediate species, and found different dependencies of HCHO consumption on H/C ratio for different temperatures. The difference is attributed to the large active energy difference between the two main HCHO consumption reactions by OH and O2. For the production part, the OH producing pathway was analysed carefully and shows H/C ratio influences OH production via H production and H abstract reactions. Our RMD simulations show that H/C ratio plays an important role in the oxidation of fuel. read less
NOT USED (high confidence) Yuan 源 Dong 董, M. Gahl, C. Zhang 张, and J. Lin, “Computational study of precision nitrogen doping on graphene nanoribbon edges,” Nanotechnology. 2017. link Times cited: 17
Abstract: Nitrogen doping in graphene is important for applications sp… read more
Abstract: Nitrogen doping in graphene is important for applications spanning from electronics to metal-free electrocatalysts. Despite much experimental study, limited theoretic work has been done in understanding the mechanism of the doping process, especially from a precision perspective. Herein, we present a computational study on precision nitrogen doping on edges of graphene nanoribbons (GNRs) by combining molecular dynamics (MD) simulation at a time scale of 40 ns and density function theory (DFT) calculation. In the MD study both ammonia and acetonitrile were used as nitrogen sources. MD results revealed that the ammonia produces almost all amine-type dopants, while the acetonitrile produces a considerable amount of pyrrolic and pyridinic nitrogen dopants which are beneficial to electronics and electrocatalysts. Results also show that the concentration of pyrrolic and pyridinic dopants can be precisely controlled by the edge geometries of the GNRs. Furthermore, DFT calculation illustrated the reaction mechanism in these different types of the GNRs when using acetonitrile as the nitrogen source. The calculated energies in different reaction stages indicate the stability of dopants on various GNRs, agreeing well with the MD results. The disclosed mechanism of controllable nitrogen doping on the edges of the GNRs would provide guidance to experimental realization, paving new routes to widespread applications. read less
NOT USED (high confidence) M. Wood, D. Kittell, C. Yarrington, and A. Thompson, “Multiscale modeling of shock wave localization in porous energetic material,” Physical Review B. 2017. link Times cited: 57
Abstract: Shock wave interactions with defects, such as pores, are kno… read more
Abstract: Shock wave interactions with defects, such as pores, are known to play a key role in the chemical initiation of energetic materials. The shock response of hexanitrostilbene is studied through a combination of large scale reactive molecular dynamics and mesoscale hydrodynamic simulations. In order to extend our simulation capability at the mesoscale to include weak shock conditions (< 6 GPa), atomistic simulations of pore collapse are used to define a strain rate dependent strength model. Comparing these simulation methods allows us to impose physically-reasonable constraints on the mesoscale model parameters. In doing so, we have been able to study shock waves interacting with pores as a function of this viscoplastic material response. We find that the pore collapse behavior of weak shocks is characteristically different to that of strong shocks. read less
NOT USED (high confidence) C. Chen, L. Zhao, J. Wang, and S. Lin, “Reactive Molecular Dynamics Simulations of Biomass Pyrolysis and Combustion under Various Oxidative and Humidity Environments,” Industrial & Engineering Chemistry Research. 2017. link Times cited: 49
Abstract: Biomass, as a renewable carbon neutral energy source with ab… read more
Abstract: Biomass, as a renewable carbon neutral energy source with abundant reserves, is a good candidate for future energy supplies. In this paper, a simplified biomass model composed of cellulose, hemicellulose, and lignin, described by a carefully selected reactive force field (ReaxFF), is investigated using molecular dynamics (MD) simulations. The pyrolysis and combustion processes of the biomass under different temperatures and oxidative and humidity conditions, are studied. We find that the individual products from the pyrolysis of the three biomass components are similar, including H2O, H2, CO, CO2, and small organic molecules. The calculated activation energies for C–C bond dissociation are 34.53, 26.08, and 16.23 kJ mol–1, respectively, for cellulose, hemicellulose, and lignin, consistent with the trend in experiments. Interestingly, light tar (C5–13) production reaches a maximum under intermediate temperatures, which could be further explored to optimize the production of light tar as liquid fuels. Compa... read less
NOT USED (high confidence) J. Yuan et al., “Shock response of 1,3,5-trinitroperhydro-1,3,5-triazine (RDX): The C-N bond scission studied by molecular dynamics simulations,” Journal of Applied Physics. 2017. link Times cited: 9
Abstract: The shock response has a great influence on the design, synt… read more
Abstract: The shock response has a great influence on the design, synthesis, and application of energetic materials in both industrial and military areas. Therefore, the initial decomposition mechanism of bond scission at the atomistic level of condensed-phase α-RDX under shock loading has been studied based on quantum molecular dynamics simulations in combination with a multi-scale shock technique. First, based on the frontier molecular orbital theory, our calculated result shows that the N-NO2 bond is the weakest bond in the α-RDX molecule in the ground state, which may be the initial bond for pyrolysis. Second, the changes of bonds under shock loading are investigated by the changes of structures, kinetic bond lengths, and Laplacian bond orders during the simulation. Also, the variation of thermodynamic properties with time in shocked α-RDX at 10 km/s along the lattice vector a for a timescale of up to 3.5 ps is presented. By analyzing the detailed structural changes of RDX under shock loading, we find that the ... read less
NOT USED (high confidence) M. Islam and A. Strachan, “Decomposition and Reaction of Polyvinyl Nitrate under Shock and Thermal Loading: A ReaxFF Reactive Molecular Dynamics Study,” Journal of Physical Chemistry C. 2017. link Times cited: 39
Abstract: We use molecular dynamics (MD) simulations with the reactive… read more
Abstract: We use molecular dynamics (MD) simulations with the reactive force field ReaxFF to investigate the response of polyvinyl nitrate (PVN), a high-energy polymer, to shock loading using the Hugoniostat technique. We compare predictions from three widely used ReaxFF versions, and in all cases, we observe shock-induced, volume-increasing exothermic reactions following a short induction time for strong enough insults. The three models predict NO2 dissociation to be the first chemical, and relatively similar final product populations; however, we find significant differences in intermediate populations indicating different reaction mechanisms due to discrepancies in the relative stability of various intermediate fragments. A time-resolved spectral analysis of the reactive MD trajectories enables the first direct comparison of shock-induced chemistry between atomistic simulations and experiments; specifically, ultrafast spectroscopy on laser shocked samples. The results from one of the ReaxFF versions are in excel... read less
NOT USED (high confidence) H. Chu, L. Cao, X. Peng, and G. Li, “Polarizable force field development for lipids and their efficient applications in membrane proteins,” Wiley Interdisciplinary Reviews: Computational Molecular Science. 2017. link Times cited: 9
Abstract: Polarizable force fields have been developed due to the intr… read more
Abstract: Polarizable force fields have been developed due to the intrinsic problem of additive force fields in modeling electrostatic interactions. Because of the capability to accurately describe the behavior of systems with significant changes in their electrostatic environments, polarizable force fields might be a decent tool to study membrane‐related systems, such as lipid bilayers, though not so much progresses have been made. In this overview article we described the developments of a variety of polarizable force fields, including the corresponding theories, benchmark examples, and more specifically we were focused on the applications on lipid membranes. WIREs Comput Mol Sci 2017, 7:e1312. doi: 10.1002/wcms.1312 read less
NOT USED (high confidence) N. Wang, J.-hua Peng, A.-min Pang, T. He, F. Du, and A. Jaramillo-Botero, “Thermodynamic Simulation of the RDX–Aluminum Interface Using ReaxFF Molecular Dynamics,” Journal of Physical Chemistry C. 2017. link Times cited: 39
Abstract: We use reactive molecular dynamics (RMD) simulations to stud… read more
Abstract: We use reactive molecular dynamics (RMD) simulations to study the interface between cyclotrimethylene trinitramine (RDX) and aluminum (Al) with different oxide layers to elucidate the effect of nanosized Al on thermal decomposition of RDX. A published ReaxFF force field for C/H/N/O elements was retrained to incorporate Al interactions and then used in RMD simulations to characterize compound energetic materials. We find that the predicted adsorption energies for RDX on the Al(111) surface and the apparent activation energies of RDX and RDX/Al are in agreement with ab initio calculations. The Al(111) surface-assisted decomposition of RDX occurs spontaneously without potential barriers, but the decomposition rate becomes slow when compared with that for RDX powder. We also find that the Al(111) surface with an oxide layer (Al oxide) slightly increases the potential barriers for decomposition of RDX molecules, while α-Al2O3(0001) retards thermal decomposition of RDX, due to the changes in thermal decompositi... read less
NOT USED (high confidence) F. J. Domínguez-Gutiérrez and P. Krstic, “Chemical sputtering of boronized and oxidized carbon surfaces irradiated by low-energy deuterium atoms,” Journal of Applied Physics. 2017. link Times cited: 10
Abstract: We use molecular dynamics to study the chemical sputtering o… read more
Abstract: We use molecular dynamics to study the chemical sputtering of boronized and oxidized amorphous carbon surfaces by deuterium irradiation in the range of impact energies of 5–30 eV. We report the sputtering yield as well as mass, energy, and angular spectra of ejected atoms and molecules of both virgin and deuterium saturated BCO surfaces and compare them with our data for a deuterated BC surface and existing theoretical and experimental results for amorphous C:D surfaces. Boron significantly suppresses the erosion of carbon, while the presence of oxygen results in further suppression. read less
NOT USED (high confidence) S. S. Choi and C. E. Son, “Analytical method for the estimation of transfer and detection efficiencies of solid state explosives using ion mobility spectrometry and smear matrix,” Analytical Methods. 2017. link Times cited: 11
Abstract: Smearing method can be used as a simple and convenient sampl… read more
Abstract: Smearing method can be used as a simple and convenient sampling method for detection of trace explosives using a portable ion mobility spectrometer (IMS) in the field such as for security checks. A testing method for efficient estimation of solid state explosives transfer and IMS detection using the smear matrix was established. Furthermore, 2,4,6-trinitrotoluene (TNT) and 1,3,5-trinitro-1,3,5-triazacyclohexane (RDX) were used as model explosives, and stainless steel mesh, cellulose paper, and cotton fabric were employed as smear matrices. Samples of solid state explosives were prepared by dropping the solutions on a poly(tetrafluoroethylene) (PTFE) sheet, and the solid state explosives deposited on the PTFE sheet were then transferred to the smear matrix using a stainless steel roller. The explosive material transferred to the smear matrix was directly measured using IMS. The minimum amounts of the explosives deposited on the PTFE sheet to detect with IMS (amount of threshold, AOT) were analyzed. The AOT(TNT)s (500 ng) were nearly the same irrespective of the smear matrix types, while the AOT(RDX)s (100–500 ng) varied according to the smear matrix types. Order of the transfer and detection efficiencies of TNT and RDX was cotton fabric > stainless steel mesh > cellulose paper. Difference in the AOTs was explained by the abilities for trapping and desorption of explosive materials. read less
NOT USED (high confidence) B. Mercer, E. Zywicz, and P. Papadopoulos, “Molecular dynamics modeling of PPTA crystallite mechanical properties in the presence of defects,” Polymer. 2017. link Times cited: 29
NOT USED (high confidence) J. H. Lee, J. C. Kim, W. C. Jeon, S. Cho, and S. Kwak, “Explosion Study of Nitromethane Confined in Carbon Nanotube Nanocontainer via Reactive Molecular Dynamics,” Journal of Physical Chemistry C. 2017. link Times cited: 9
Abstract: Explosion dynamics of confined nitromethane (NM) fluid has b… read more
Abstract: Explosion dynamics of confined nitromethane (NM) fluid has been investigated by using nonequilibrium reactive molecular dynamics. For the confinement, NM was encapsulated into a nanocontainer, which is the capped (20, 20) armchair carbon nanotube (CNT). After thermal energy was injected into confined NM at various densities, the nanobomb consisting of NM and CNT was fully decomposed including bursting phenomena. We found that the time for explosion was reduced as density and initial temperature increased. While NM was being decomposed into intermediates, defects of Stone–Wales type (5–7 carbon atoms ring) or high-order rings were randomly formed at the cap and side wall of CNT. Subsequently, the intermediates functionalized carbon atoms at the defects, from which nanoholes were evolved. The CNT burst when the size of nanohole became about 8 A. Further, we demonstrated that defective CNT with vacancy exploded faster because carbon atoms at defect sites played a seed role to make nanoholes. This theoretical... read less
NOT USED (high confidence) X. Xue, Y. Ma, Q. Zeng, and C. Zhang, “Initial Decay Mechanism of the Heated CL-20/HMX Cocrystal: A Case of the Cocrystal Mediating the Thermal Stability of the Two Pure Components,” Journal of Physical Chemistry C. 2017. link Times cited: 53
Abstract: Energetic cocrystallization, by combining existing molecules… read more
Abstract: Energetic cocrystallization, by combining existing molecules together, is thought to be new strategy for creating energetic materials. Nevertheless, the underlying mechanism of its influences on properties and performances in comparison with their pure components remains unclear. The present work reveals the cocrystallization influence of a typical energetic cocrystal of CL-20/HMX on thermal stability, by ReaxFF molecular reactive dynamic simulations and kinetics calculations on the pure and cocrystals. As a result, we find that the cocrystal mediates the thermal stability of pure crystalsand this is in agreement with experimental observations. The initial decay steps in pure crystals remain still in the cocrystal, that is, the independent and intramolecular reactions of N–N bond cleavage governing the initial decay of the pure CL-20 and HMX crystals also dominate in the cocrystal of CL-20/HMX. Meanwhile, during the thermal decomposition of the cocrystal, CL-20 releases heat faster than HMX, thus the heat... read less
NOT USED (high confidence) T. Nagy and M. Meuwly, “Modelling Chemical Reactions Using Empirical Force Fields.” 2017. link Times cited: 1
Abstract: Chemical reactions involve bond-breaking and bond-forming pr… read more
Abstract: Chemical reactions involve bond-breaking and bond-forming processes and are fundamental in chemistry and the life sciences in general. In many cases, mechanistic aspects of the reactions (“which reaction partners interact at which time with each other”) are of interest. However, many atomistic aspects in bond-breaking and bond-forming processes remain elusive by considering experimental data alone because “the reaction” itself is a transient process. The transition state is unstable and short-lived. Thus, the most interesting regions along a reaction path can not be investigated experimentally in a direct fashion. To shed light on such questions, theoretical and computational work has become invaluable to experimental efforts in understanding particular reaction schemes. The computational investigation of a chemical or biological system requiresmodels to compute the total energy of the systemunder investigation.There are two fundamentally different concepts to do that: either by solving the electronic Schrödinger equation, or by assuming a suitably defined empirical potential energy function. The first approach has been refined to a degree that allows one to carry out calculations with “chemical accuracy” – that is, accuracies for relative energies within 1 kcal/mol for the chemically bonded region and less accurately for transition state regions.Most importantly, a quantum chemical calculation makes no assumption on the bonding pattern in the molecule and is ideally suited to answer the question which atoms are bonded to one another for a particular relative arrangement of the atoms. To obtain realistic reaction profiles it is, however, necessary to carry out calculations at a sufficiently high level of theory, particularly in the region of the transition state. Through statistical mechanics and assuming idealized models of molecular motion such as rigid rotor or harmonic oscillator, average internal energies, enthalpies, and by including entropic effects, also free energies can be calculated. However, although such computations are by now standard, they can realistically and routinely only be carried out for systems including several tens of heavy atoms, that is, small systems in the gas phase.This is due to the N3 scaling of the secular determinants that need to be diagonalized, where N is the number of basis functions. Alternative approaches to solving the electronic Schrödinger equation have been developed and matured to similar degrees. London’s work on the H +H2 reaction for read less
NOT USED (high confidence) C. M. Ashraf, A. Jain, Y. Xuan, and A. V. van Duin, “ReaxFF based molecular dynamics simulations of ignition front propagation in hydrocarbon/oxygen mixtures under high temperature and pressure conditions.,” Physical chemistry chemical physics : PCCP. 2017. link Times cited: 36
Abstract: In this paper, we present the first atomistic-scale based me… read more
Abstract: In this paper, we present the first atomistic-scale based method for calculating ignition front propagation speed and hypothesize that this quantity is related to laminar flame speed. This method is based on atomistic-level molecular dynamics (MD) simulations with the ReaxFF reactive force field. Results reported in this study are for supercritical (P = 55 MPa and Tu = 1800 K) combustion of hydrocarbons as elevated pressure and temperature are required to accelerate the dynamics for reactive MD simulations. These simulations are performed for different types of hydrocarbons, including alkyne, alkane, and aromatic, and are able to successfully reproduce the experimental trend of reactivity of these hydrocarbons. Moreover, our results indicate that the ignition front propagation speed under supercritical conditions has a strong dependence on equivalence ratio, similar to experimentally measured flame speeds at lower temperatures and pressures which supports our hypothesis that ignition front speed is a related quantity to laminar flame speed. In addition, comparisons between results obtained from ReaxFF simulation and continuum simulations performed under similar conditions show good qualitative, and reasonable quantitative agreement. This demonstrates that ReaxFF based MD-simulations are a promising tool to study flame speed/ignition front speed in supercritical hydrocarbon combustion. read less
NOT USED (high confidence) S. Mun, A. Bowman, S. Nouranian, S. Gwaltney, M. Baskes, and M. Horstemeyer, “Interatomic Potential for Hydrocarbons on the Basis of the Modified Embedded-Atom Method with Bond Order (MEAM-BO).,” The journal of physical chemistry. A. 2017. link Times cited: 18
Abstract: In this paper, we develop a new modified embedded atom metho… read more
Abstract: In this paper, we develop a new modified embedded atom method (MEAM) potential that includes the bond order (MEAM-BO) to describe the energetics of unsaturated hydrocarbons (double and triple carbon bonds) and also develop improved parameters for saturated hydrocarbons from those of our previous work. Such quantities like bond lengths, bond angles, and atomization energies at 0 K, dimer molecule interactions, rotational barriers, and the pressure-volume-temperature relationships of dense systems of small molecules give a comparable or more accurate property relative to experimental and first-principles data than the classical reactive force fields REBO and ReaxFF. Our extension of the MEAM potential for unsaturated hydrocarbons (MEAM-BO) is a step toward developing more reliable and accurate polymer simulations with their associated structure-property relationships, such as reactive multicomponent (organic/metal) systems, polymer-metal interfaces, and nanocomposites. When the constants for the BO are zero, MEAM-BO reduces to the original MEAM potential. As such, this MEAM-BO potential describing the interaction of organic materials with metals within the same MEAM formalism is a significant advancement for computational materials science. read less
NOT USED (high confidence) L. Koziol, L. Fried, and N. Goldman, “Using Force Matching To Determine Reactive Force Fields for Water under Extreme Thermodynamic Conditions.,” Journal of chemical theory and computation. 2017. link Times cited: 24
Abstract: We present a method for the creation of classical force fiel… read more
Abstract: We present a method for the creation of classical force fields for water under dissociative thermodynamic conditions by force matching to molecular dynamics trajectories from Kohn-Sham density functional theory (DFT). We apply our method to liquid water under dissociative conditions, where molecular lifetimes are less than 1 ps, and superionic water, where hydrogen ions diffuse at liquid-like rates through an oxygen lattice. We find that, in general, our new models are capable of accurately reproducing the structural and dynamic properties computed from DFT, as well as the molecular concentrations and lifetimes. Overall, our force-matching approach presents a relatively simple way to create classical reactive force fields for a single thermodynamic state point that largely retains the accuracy of DFT while having the potential to access experimental time and length scales. read less
NOT USED (high confidence) Y. Long and J. Chen, “Theoretical study of the reaction kinetics and the detonation wave profile for 1,3,5-triamino-2,4,6-trinitrobenzene,” Journal of Applied Physics. 2016. link Times cited: 8
Abstract: We simulate the reaction process of 1,3,5-triamino-2,4,6-tri… read more
Abstract: We simulate the reaction process of 1,3,5-triamino-2,4,6-trinitrobenzene in wide temperature and pressure ranges by molecular dynamics and evaluate the intermediate molecules, chemical reaction rates, and Hugoniot relations. Based on them, the leading shock wave, fast reaction zone, Chapman-Jouguet state, and slow reaction zone under detonation are investigated by different theoretical methods. A complete structure of the detonation wave is obtained. The calculated detonation velocity, detonation pressure, detonation products, and the length of the reaction zone are in agreement with the experiments and others' calculations. We find that some intermediate molecules play an important role in determining the reaction path of explosives but just remain a little after detonation, such as H2 and NH3. read less
NOT USED (high confidence) Y. Long and J. Chen, “The force-field derivation and application of explosive/additive interfaces,” Modelling and Simulation in Materials Science and Engineering. 2016. link Times cited: 2
Abstract: The inter-molecular force-field across RDX/(paraffin, fluoro… read more
Abstract: The inter-molecular force-field across RDX/(paraffin, fluoropolymer) interfaces are derived from first-principles calculated energies under the GGA+vdW functional. Based on the force-field, the polycrystal structures of mixture explosives are obtained, and a set of thermodynamic properties are calculated, including the elastic constants, thermal expansion coefficient, heat capacity, isothermal curve and the Hugoniot curve. The results are in good agreement with the available experiments, and provide a reasonable prediction about the properties of plastic bonded explosives. We find that the thermal expansion coefficient of a multi-component explosive is not only determined by the properties of the components, but is also affected by the thermal stress at the explosive/additive interfaces. read less
NOT USED (high confidence) Z.-H. He, J. Chen, Q. Wu, and G. Ji, “Special catalytic effects of intermediate-water for rapid shock initiation of β-HMX,” RSC Advances. 2016. link Times cited: 8
Abstract: Quantum-based multiscale calculations were carried out to re… read more
Abstract: Quantum-based multiscale calculations were carried out to reveal the rapid shock initiation mechanism of β-HMX (octahydro-1,3,5,7-tetranitro-1,3,5,7-tetrazocine). Tracking the dissociation process of HMX, we discovered a special catalytic action of intermediate-water. During early decomposition of HMX, water and its derivative (·OH) acted as oxidizer, dominated the carbon oxidation reaction, and promoted the oxygen-transport from nitrogen to carbon. Water monomer and its small polymers efficiently transferred proton and hydroxyl moieties between reaction centers, also contributing to the acceleration of the carbon oxidation reaction. The carbon oxidation significantly reduced the dissociation energy barrier of C–N bonds, causing fast and deep cracking of HMX. This water catalytic mechanism may contribute to the illustration of the intrinsic difference in reaction properties between sensitive and insensitive explosives, and shed light on understanding the rapid shock initiation process. read less
NOT USED (high confidence) X. Xue, Y. Wen, and C. Zhang, “Early Decay Mechanism of Shocked ε-CL-20: A Molecular Dynamics Simulation Study,” Journal of Physical Chemistry C. 2016. link Times cited: 41
Abstract: e-2,4,6,8,10,12-Hexanitro-2,4,6,8,10,12-hexaazaisowurtzitane… read more
Abstract: e-2,4,6,8,10,12-Hexanitro-2,4,6,8,10,12-hexaazaisowurtzitane (CL-20) is currently the most powerful explosive commercially available. Nevertheless, the early decay events of shocked e-CL-20 still remain unclear. We perform quantum based self-consistent charge density-functional tight-binding molecular dynamics simulations, in combination with the multiscale shock simulation technique, to reveal the events with four specified shock velocities (Us) of 8 to 11 km/s. We find that the temperature and pressure increases and that the volume reduction is enhanced with increasing shock strength. The ring opening is observed to trigger molecular decay at all four shock conditions; while the sufficient NO2 fission is observed at Us = 8 and 9 km/s, and strongly inhibited at Us = 10 and 11 km/s. Moreover, the evolution of main chemical species, such as active intermediates, stable products, and clusters, is strongly dependent on the shock strength. NO2 and H are dominant in the primary intermediates, responsible for w... read less
NOT USED (high confidence) R. Chaudret, A. Bick, and X. Krokidis, “Theoretical Modeling of Thermal Decomposition of Methylnaphthalene Derivatives: Influence of Substituents,” Energy & Fuels. 2016. link Times cited: 6
Abstract: The kinetics and thermodynamics of thermal decomposition of … read more
Abstract: The kinetics and thermodynamics of thermal decomposition of naphthalene and several mono- and dimethylnaphathalene derivatives have been established through reactive molecular dynamics simulations using ReaxFF. These results were compared to previous theoretical and experimental studies and also compared to density functional theory calculation results. This work demonstrates that the kinetics and thermodynamics of the initial activation reaction is directly impacted by the position and number of methyl substituents. Subsequently, the activated molecules react to form either small organic or large char molecules. The char formation mechanism is shown to occur in three steps: activation, dimerization/trimerization, and condensation. Finally, temperature effects on the char formation reaction were also studied. read less
NOT USED (high confidence) Y. Dong, S. C. Rismiller, and J. Lin, “Molecular dynamic simulation of layered graphene clusters formation from polyimides under extreme conditions,” Carbon. 2016. link Times cited: 90
NOT USED (high confidence) X. Hu, N. Chen, and W. Li, “A method for fast safety screening of explosives in terms of crystal packing and molecular stability,” Journal of Molecular Modeling. 2016. link Times cited: 5
NOT USED (high confidence) K. Banlusan and A. Strachan, “Shockwave Energy Dissipation in Metal–Organic Framework MOF-5,” Journal of Physical Chemistry C. 2016. link Times cited: 27
Abstract: We investigate the response of the metal–organic framework M… read more
Abstract: We investigate the response of the metal–organic framework MOF-5 to shock loading along various crystallographic orientations using molecular dynamics simulations with the reactive force field ReaxFF. The dynamical compressive load leads to the volumetric collapse of the open structure of MOF-5. A two-wave structure forms with a pore-collapse wave following a leading elastic precursor that propagates at a higher speed. Interestingly, the propagating pore-collapse wave weakens the elastic precursor indicating possible applications of these materials for shockwave dissipation. Within the two-wave regime, an increase in piston velocity leads to an increase in the velocity of the pore-collapse wave, but the pressure and velocity of the elastic precursor remain unchanged. For piston velocities between 2 and 3 km/s (depending on orientation) the pore-collapse wave catches up with the elastic precursor leading to an overdriven regime. The simulations yield insight into the molecular process of pore collapse and ... read less
NOT USED (high confidence) H.-ping Cheng, Z. Huang, and T.-nan Chen, “Electronic structure and optical properties of α-RDX crystal under pressure from first-principles calculations,” Molecular Physics. 2016. link Times cited: 1
Abstract: ABSTRACT In this study, we investigate the electronic struct… read more
Abstract: ABSTRACT In this study, we investigate the electronic structure and optical properties of α-RDX crystal under pressure from 0 to 4 GPa using first-principles calculations based on the density functional theory. When pressure increases, the calculations figure out that the band gap linearly decreases, the peaks of the total density of states are lowered, the valence bandwidths broadened and the conduction bandwidths compressed. The results also show that the optical properties including dielectric function, absorption spectrum, refractive index, reflectivity and energy-loss function respond to compression, that is, the intensity of the maximal peak of absorption coefficient increases from 7.07 × 104 to 7.98 × 104 cm−1 with corresponding photon energy from 5.10 to 5.15 eV, the static refractive index increases from 1.23 to 1.30, the intensity of the maximal peak of reflectivity function increases from 0.24 to 0.32 accompanied by its photon energy from 5.74 to 5.97 eV, and the plasma resonance energy and its intensity increases from 5.92 to 6.20 eV and 3.81 to 5.13, respectively. read less
NOT USED (high confidence) M. Wood and A. Strachan, “Nonequilibrium Reaction Kinetics in Molecular Solids,” Journal of Physical Chemistry C. 2016. link Times cited: 12
Abstract: We explore the possibility of nonstatistical chemical reacti… read more
Abstract: We explore the possibility of nonstatistical chemical reactions in condensed-phase energetic materials via reactive molecular dynamics (MD) simulations. We characterize the response of nitromethane [CH3NO2], HMX [cyclic (CH2-NNO2)4], and PETN [C-(CH2-ONO2)4] to different types of insults: electric fields of various frequencies and strengths and direct heating at various rates. We find that nonequilibrium states can be created for short time scales when energy input targets specific vibrations through the electric fields and that equilibration eventually occurs even while the insults remain present. Interestingly, for strong fields these relaxation time scales are comparable to those of the initial chemical decomposition of the molecules. NM decomposes predominantly via bimolecular reactions, and while insults targeting specific modes lead to strong nonequilibrium states, they do not affect the kinetics associated with decomposition. PETN decomposes via the unimolecular formation of NO2 and, quite interest... read less
NOT USED (high confidence) A. Rahnamoun and A. Duin, “Study of ice cluster impacts on amorphous silica using the ReaxFF reactive force field molecular dynamics simulation method,” Journal of Applied Physics. 2016. link Times cited: 5
Abstract: We study the dynamics of the collisions between amorphous si… read more
Abstract: We study the dynamics of the collisions between amorphous silica structures and amorphous and crystal ice clusters with impact velocities of 1 km/s, 4 km/s, and 7 km/s using the ReaxFF reactive molecular dynamics simulation method. The initial ice clusters consist of 150 water molecules for the amorphous ice cluster and 128 water molecules for the crystal ice cluster. The ice clusters are collided on the surface of amorphous fully oxidized and suboxide silica. These simulations show that at 1 km/s impact velocities, all the ice clusters accumulate on the surface and at 4 km/s and 7 km/s impact velocities, some of the ice cluster molecules bounce back from the surface. At 4 km/s and 7 km/s impact velocities, few of the water molecules dissociations are observed. The effect of the second ice cluster impacts on the surfaces which are fully covered with ice, on the mass loss/accumulation is studied. These studies show that at 1 km/s impacts, the entire ice cluster accumulates on the surface at both first and ... read less
NOT USED (high confidence) Y. Han, D. D. Jiang, J. Zhang, W. Li, Z. Gan, and J. Gu, “Development, applications and challenges of ReaxFF reactive force field in molecular simulations,” Frontiers of Chemical Science and Engineering. 2016. link Times cited: 87
NOT USED (high confidence) S. Naserifar, S. Zybin, C. Ye, and W. Goddard, “Prediction of structures and properties of 2,4,6-triamino-1,3,5-triazine-1,3,5-trioxide (MTO) and 2,4,6-trinitro-1,3,5-triazine-1,3,5-trioxide (MTO3N) green energetic materials from DFT and ReaxFF molecular modeling,” Journal of Materials Chemistry. 2016. link Times cited: 13
Abstract: 2,4,6-Triamino-1,3,5-triazine-1,3,5-trioxide (MTO) and 2,4,6… read more
Abstract: 2,4,6-Triamino-1,3,5-triazine-1,3,5-trioxide (MTO) and 2,4,6-trinitro-1,3,5-triazine-1,3,5-trioxide (MTO3N) were suggested by Klapotke et al. as candidates for green high energy density materials (HEDM), but a successful synthesis has not yet been reported. In order to predict the properties of these systems, we used quantum mechanics (PBE flavor of density functional theory) to predict the most stable conformations of MTO and MTO3N and their optimum packing into the most stable crystal structures. We found that MTO has the P21 space-group with a density of ρ = 1.92 g cm−3 while MTO3N has the P21/c space-group with a density of ρ = 2.10 g cm−3. The heats of reaction (ΔHrxn) were computed to be 1036 kcal kg−1 for MTO, 1412 kcal kg−1 for MTO3N, and 1653 kcal kg−1 for a mixture of them. These properties are comparable to those of such other useful energetic materials as RDX (ρ = 1.80 g cm−3, ΔHrxn = 1266 kcal kg−1), HMX, and PETN, making MTO and MTO3N excellent candidates for environmentally friendly HEDMs. In addition, we predicted the stability of –NH2, –NO, and –NO2 groups in water solution. We also show that the ReaxFF-lg reactive FF leads to an accurate description of the structural properties of MTO and MTO3N crystals making it practical to carry out large-scale reactive molecular dynamics simulations practical for these systems to determine the sensitivity and performance (CJ point calculation and velocity) under shear, shock, and thermal loads. read less
NOT USED (high confidence) F. Guo, H. Zhang, H. Hu, X. Cheng, and L.-Y. Zhang, “Hugoniot curve calculation of nitromethane decomposition mixtures: A reactive force field molecular dynamics approach*,” Chinese Physics B. 2015. link Times cited: 5
Abstract: We investigate the Hugoniot curve, shock–particle velocity r… read more
Abstract: We investigate the Hugoniot curve, shock–particle velocity relations, and Chapman–Jouguet conditions of the hot dense system through molecular dynamics(MD) simulations. The detailed pathways from crystal nitromethane to reacted state by shock compression are simulated. The phase transition of N2 and CO mixture is found at about 10 GPa, and the main reason is that the dissociation of the C–O bond and the formation of C–C bond start at 10.0–11.0 GPa. The unreacted state simulations of nitromethane are consistent with shock Hugoniot data. The complete pathway from unreacted to reacted state is discussed. Through chemical species analysis, we find that the C–N bond breaking is the main event of the shock-induced nitromethane decomposition. read less
NOT USED (high confidence) M. R. Weismiller, C. Junkermeier, M. F. Russo, M. R. Salazar, D. Bedrov, and A. V. van Duin, “ReaxFF molecular dynamics simulations of intermediate species in dicyanamide anion and nitric acid hypergolic combustion,” Modelling and Simulation in Materials Science and Engineering. 2015. link Times cited: 6
Abstract: Ionic liquids based on the dicyanamide anion (DCA) are of in… read more
Abstract: Ionic liquids based on the dicyanamide anion (DCA) are of interest as replacements for current hypergolic fuels, which are highly toxic. To better understand the reaction dynamics of these ionic liquid fuels, this study reports the results of molecular dynamics simulations performed for two predicted intermediate compounds in DCA-based ionic liquids/nitric acid (HNO3) combustion, i.e. protonated DCA (DCAH) and nitro-dicyanamide-carbonyl (NDC). Calculations were performed using a ReaxFF reactive force field. Single component simulations show that neat NDC undergo exothermic decomposition and ignition. Simulations with HNO3 were performed at both a low (0.25 g ml−1) and high (1.00 g ml−1) densities, to investigate the reaction in a dense vapor and liquid phase, respectively. Both DCAH and NDC react hypergolically with HNO3, and increased density led to shorter times for the onset of thermal runaway. Contrary to a proposed mechanism for DCA combustion, neither DCAH nor NDC are converted to 1,5-Dinitrobiuret (DNB) before thermal runaway. Details of reaction pathways for these processes are discussed. read less
NOT USED (high confidence) B. D. Jensen, K. Wise, and G. Odegard, “Simulation of the Elastic and Ultimate Tensile Properties of Diamond, Graphene, Carbon Nanotubes, and Amorphous Carbon Using a Revised ReaxFF Parametrization.,” The journal of physical chemistry. A. 2015. link Times cited: 100
Abstract: In light of the enduring interest in using nanostructured ca… read more
Abstract: In light of the enduring interest in using nanostructured carbon materials as reinforcing elements in composite materials, there is a significant need for a reliable computational tool capable to predict the mechanical properties, both elastic properties and properties at the point of fracture, in large-scale atomistic simulations. A revised version of the ReaxFF reactive force field parametrization for carbon, ReaxFFC-2013, was recently published and is notable because of the inclusion of density functional theory (DFT)-derived mechanical data for diamond and graphite in the fitting set. The purpose of the present work is to assess the accuracy of this new force field for predicting the mechanical properties for several allotropes of carbon, both in the elastic regime and during fracture. The initial discussion focuses on the performance of ReaxFFC-2013 for diamond and graphene, the two carbon forms for which mechanical properties were included in the parametrization data set. After it is established that simulations conducted with the new force field yield results that agree well with DFT and experimental data for most properties of interest, its transferability to amorphous carbon and carbon nanotubes is explored. ReaxFFC-2013 is found to produce results that, for the most part, compare favorably with available experimental data for single and multiwalled nanotubes and for amorphous carbon models prepared over a range of densities. Although there is opportunity for improvement in some predicted properties, the ReaxFFC-2013 parametrization is shown to generally perform well for each form of carbon and to compare favorably with DFT and experimental data. read less
NOT USED (high confidence) Y. Zhang, X. Wang, Q. Li, R. Yang, and C. Li, “A ReaxFF Molecular Dynamics Study of the Pyrolysis Mechanism of Oleic-type Triglycerides,” Energy & Fuels. 2015. link Times cited: 39
Abstract: The reactive force field (ReaxFF) method is employed in the … read more
Abstract: The reactive force field (ReaxFF) method is employed in the molecular dynamics (MD) simulation of oleic-type triglyceride (OTG) pyrolysis for the first time. The complex pyrolysis mechanism of OTG at high temperature, especially focusing on the multichannel pyrolysis pathways of OTG and radical-related evolution mechanisms of products, is intensively investigated at the atomistic level by performing a series of ReaxFF MD simulations. On the basis of simulation trajectory analysis, we find that the initiation decomposition of OTG pyrolysis is through C–O bond fission to release the straight oleic acid radical (C18H33O2•). The decomposition of C18H33O2• radical is mainly started through multichannel pathways: the decarboxylation reaction to form long-chain hydrocarbon radical (C17H33•) and CO2, and C–C bond cleavages at α, β-C position to form hydrocarbon radicals and ester radicals. C–C bond β-scissions and conjugation reactions play important roles in the subsequent decomposition of the C18H33O2• radical.... read less
NOT USED (high confidence) X. Xue, Y. Wen, X. Long, J. Li, and C. Zhang, “Influence of Dislocations on the Shock Sensitivity of RDX: Molecular Dynamics Simulations by Reactive Force Field,” Journal of Physical Chemistry C. 2015. link Times cited: 42
Abstract: Molecular dynamics simulations of the chemical responses of … read more
Abstract: Molecular dynamics simulations of the chemical responses of shocked dislocation-contained and perfect (p) 1,3,5-trinitro-1,3,5-triazinane (RDX) crystals were performed using the ReaxFF force field combined with the multiscale shock technique. The shear dynamics of four types of dislocated RDX crystals are also modeled. The predicted mobilities of the crystals decrease in the order of (010) [001]/screw (s2) > (010) [001]/edge (e2) > (010) 1/2[100]/screw (s1) > (010)1/2[100]/edge (e1) according to their shear stress barriers, thus revealing the initial driving force required to activate a slip system. In view of the evolution of temperatures, pressures, and reactant decay rates of the shocked perfect and dislocated RDX, we confirm that shock sensitivity follows the order of e2 > e1 > s1 ≈ s2 > p. In particular, all dislocations enhance the shock sensitivity of RDX; in particular, edge dislocations enhance shock sensitivity significantly, whereas screw dislocations enhance it slightly. Shock sensitivity is n... read less
NOT USED (high confidence) C. Ye et al., “Reaction mechanism from quantum molecular dynamics for the initial thermal decomposition of 2,4,6-triamino-1,3,5-triazine-1,3,5-trioxide (MTO) and 2,4,6-trinitro-1,3,5-triazine-1,3,5-trioxide (MTO3N), promising green energetic materials,” Journal of Materials Chemistry. 2015. link Times cited: 15
Abstract: Klapotke and co-workers recently designed two new materials,… read more
Abstract: Klapotke and co-workers recently designed two new materials, 2,4,6-triamino-1,3,5-triazine-1,3,5-trioxide (MTO) and 2,4,6-trinitro-1,3,5-triazine-1,3,5-trioxide (MTO3N), envisioned as candidates for green high-energy materials. However, all attempts at synthesis have failed. In order to validate the expected properties for these systems and to determine why these materials are too unstable to synthesize, we used the PBE flavor of Density Functional Theory (DFT) to predict the crystal structures for MTO and MTO3N and then we carried out DFT molecular dynamics simulations (DFT-MD) to determine the initial reaction mechanisms for decomposition. Klapotke estimated that MTO would have a density of ρ = 1.859 g cm−3 with an estimated detonation velocity (Dv) of 8.979 km s−1, making it comparable to RDX (ρ = 1.82 g cm−3, Dv = 8.75 km s−1) and β-HMX (ρ = 1.91 g cm−3, Dv = 9.10 km s−1). His estimated impact sensitivity >30 J, make it much better than HMX (7 J) and RDX (7.5 J). Our predicted crystal structure for MTO (P2(1) space group) leads to ρ = 1.859 g cm−3, in good agreement with expectations. Our DFT-MD studies find that the first step in the decomposition of MTO is intermolecular hydrogen-transfer reaction (barrier 3.0 kcal mol−1) which is followed quickly by H2O and NO release with reaction barriers of 46.5 and 35.5 kcal mol−1. In contrast for MTO3N (P2(1)/c predicted space group), we find that the first steps are a bimolecular decomposition to release NO2 (ΔH = 44.1 kcal mol−1, ΔG = 54.7 kcal mol−1) simultaneous with unimolecular NO2 cleavage (ΔH = 59.9 and ΔG = 58.2 kcal mol−1) a unique initial reaction among EMs. These results suggest that MTO3N would be significantly more thermally stabile (barrier > 6.0 kcal mol−1 higher) than RDX and HMX, making it an excellent candidate to be insensitive new green energetic materials. However we find that MTO leads to very favorable hydrogen transfer reactions that may complicate synthesis and crystallization, making MTO3N the more promising system. read less
NOT USED (high confidence) C. Zhang, C. Zhang, Y. Ma, and X. Xue, “Imaging the C black formation by acetylene pyrolysis with molecular reactive force field simulations.,” Physical chemistry chemical physics : PCCP. 2015. link Times cited: 28
Abstract: C black is a class of substantial materials with a long hist… read more
Abstract: C black is a class of substantial materials with a long history of applications. However, apart from some descriptions of primary reactions, subsequent processes leading up to the final formation mechanism remain unclear. This mechanism is also crucial for understanding the formation of other carbonaceous materials. In this work, we visualize C black formation by acetylene pyrolysis using molecular dynamics simulations with a molecular reactive force field named ReaxFF. We find that the formation undergoes four stages: (1) chain elongation by H abstraction and polymerization of small C species, (2) chain branching, (3) cyclization and ring densification, and (4) condensed ring folding. The simulated C black particle possesses a structure of folded graphite layers, which is in good accordance with experimental observations. Cyclization and condensation are derived from fusion between neighboring chains, significantly varying from common experimental observations at relatively low temperatures that abide by the mechanism of H abstraction and C2H2 addition. Moreover, polyyne and polyene are usually found during acetylene pyrolysis, suggesting that the pyrolysis of acetylene and other hydrocarbons may be a feasible method of obtaining carbyne, a novel carbonaceous material with a high value. read less
NOT USED (high confidence) N. Onofrio and A. Strachan, “Voltage equilibration for reactive atomistic simulations of electrochemical processes.,” The Journal of chemical physics. 2015. link Times cited: 37
Abstract: We introduce electrochemical dynamics with implicit degrees … read more
Abstract: We introduce electrochemical dynamics with implicit degrees of freedom (EChemDID), a model to describe electrochemical driving force in reactive molecular dynamics simulations. The method describes the equilibration of external electrochemical potentials (voltage) within metallic structures and their effect on the self-consistent partial atomic charges used in reactive molecular dynamics. An additional variable assigned to each atom denotes the local potential in its vicinity and we use fictitious, but computationally convenient, dynamics to describe its equilibration within connected metallic structures on-the-fly during the molecular dynamics simulation. This local electrostatic potential is used to dynamically modify the atomic electronegativities used to compute partial atomic changes via charge equilibration. Validation tests show that the method provides an accurate description of the electric fields generated by the applied voltage and the driving force for electrochemical reactions. We demonstrate EChemDID via simulations of the operation of electrochemical metallization cells. The simulations predict the switching of the device between a high-resistance to a low-resistance state as a conductive metallic bridge is formed and resistive currents that can be compared with experimental measurements. In addition to applications in nanoelectronics, EChemDID could be useful to model electrochemical energy conversion devices. read less
NOT USED (high confidence) E. Antillon and A. Strachan, “Mesoscale simulations of shockwave energy dissipation via chemical reactions.,” The Journal of chemical physics. 2015. link Times cited: 16
Abstract: We use a particle-based mesoscale model that incorporates ch… read more
Abstract: We use a particle-based mesoscale model that incorporates chemical reactions at a coarse-grained level to study the response of materials that undergo volume-reducing chemical reactions under shockwave-loading conditions. We find that such chemical reactions can attenuate the shockwave and characterize how the parameters of the chemical model affect this behavior. The simulations show that the magnitude of the volume collapse and velocity at which the chemistry propagates are critical to weaken the shock, whereas the energetics in the reactions play only a minor role. Shock loading results in transient states where the material is away from local equilibrium and, interestingly, chemical reactions can nucleate under such non-equilibrium states. Thus, the timescales for equilibration between the various degrees of freedom in the material affect the shock-induced chemistry and its ability to attenuate the propagating shock. read less
NOT USED (high confidence) J. Larentzos, B. Rice, E. Byrd, N. S. Weingarten, and J. Lill, “Parameterizing complex reactive force fields using multiple objective evolutionary strategies (MOES). Part 1: ReaxFF models for cyclotrimethylene trinitramine (RDX) and 1,1-diamino-2,2-dinitroethene (FOX-7).,” Journal of chemical theory and computation. 2015. link Times cited: 42
Abstract: ReaxFF (van Duin, A.C.T.; Dasgupta, S.; Lorant, F.; Goddard, W.A. J. Phys. Chem. A, 2001, 105, 9396-9409) reactive potentials are parametrized for cyclotrimethylene trinitramine (RDX) and 1,1-diamino-2,2-dinitroethene (FOX-7) in a novel application combining data envelopment analysis and a modern self-adaptive evolutionary algorithm to optimize multiple objectives simultaneously and map the entire family of solutions. In order to correct the poor crystallographic parameters predicted by ReaxFF using its base parametrization (Strachan, A.; van Duin, A. C. T.; Chakraborty, D.; Dasgupta S.; Goddard, W. A. Phys. Rev. Lett., 2003, 91, 098301), we augmented the existing training set data used for parametrization with additional (SAPT)DFT calculations of RDX and FOX-7 dimer interactions. By adjusting a small subset of the ReaxFF parameters that govern long-range interactions, the evolutionary algorithm approach converges on a family of solutions that best describe crystallographic parameters through simultaneous optimization of the objective functions. Molecular dynamics calculations of RDX and FOX-7 are conducted to assess the quality of the force fields, resulting in parametrizations that improve the overall prediction of the crystal structures. read less
NOT USED (high confidence) T. Zhou, Y.-geng Zhang, J. Lou, H. Song, and F. Huang, “A reactive molecular dynamics study on the anisotropic sensitivity in single crystal α-cyclotetramethylene tetranitramine,” RSC Advances. 2015. link Times cited: 4
Abstract: The anisotropic shock sensitivity in single crystal α-cyclot… read more
Abstract: The anisotropic shock sensitivity in single crystal α-cyclotetramethylene tetranitramine (α-HMX) was investigated using the compress-shear reactive dynamics (CS-RD) computational protocol. Anisotropy in thermo-mechanical and chemical responses is found by measuring shear stress, energy, temperature, and chemical reactions during the dynamical process for shock directions perpendicular to the (010), (001), (100), (110), (011), (111), and (101) planes. We suggest that the internal energy accumulated within the duration of the surmounting shear stress barrier can be used as a useful criterion to distinguish the anisotropic sensitivity among various shock orientations. Accordingly, the α-HMX single crystal is predicted to be sensitive for the shock normal to the (010) plane, is intermediate to the (001) plane, and is insensitive to the (100), (110), (011), (111), and (101) planes. The molecular origin of the anisotropic sensitivity is considered to be the different intermolecular steric arrangements on the two sides of the slip plane induced by shock compression along various orientations. The shear deformation induced by shock compression along sensitive directions encounters strong intermolecular contact and has little intermolecular free space for geometry relaxation when molecules collide, leading to a high shear stress barrier and energy accumulation, which benefit a temperature increase and initial chemical bond dissociation that trigger further reactions. This validation of CS-RD indicates that this approach would be valuable in examining the anisotropic sensitivity of new energetic crystals and in evaluating which one would be least sensitive. read less
NOT USED (high confidence) T. L. Jensen, J. Moxnes, E. Unneberg, and O. Dullum, “Calculation of Decomposition Products from Components of Gunpowder by using ReaxFF Reactive Force Field Molecular Dynamics and Thermodynamic Calculations of Equilibrium Composition,” Propellants, Explosives, Pyrotechnics. 2014. link Times cited: 8
Abstract: Jensen, Tomas Lunde; Moxnes, John Fredrik; Unneberg, Erik; D… read more
Abstract: Jensen, Tomas Lunde; Moxnes, John Fredrik; Unneberg, Erik; Dullum, Ove.
Calculation of decomposition products from components of gunpowder by using ReaxFF reactive force field molecular dynamics and thermodynamic calculations of equilibrium composition. Propellants, explosives, pyrotechnics 2014 ;Volum 39.(6) s. 830-837 read less
NOT USED (high confidence) Y. Li, Y. Li, R. Kalia, A. Nakano, K. Nomura, and P. Vashishta, “Multistage reaction pathways in detonating high explosives,” Applied Physics Letters. 2014. link Times cited: 23
Abstract: Atomistic mechanisms underlying the reaction time and interm… read more
Abstract: Atomistic mechanisms underlying the reaction time and intermediate reaction products of detonating high explosives far from equilibrium have been elusive. This is because detonation is one of the hardest multiscale physics problems, in which diverse length and time scales play important roles. Here, large spatiotemporal-scale reactive molecular dynamics simulations validated by quantum molecular dynamics simulations reveal a two-stage reaction mechanism during the detonation of cyclotrimethylenetrinitramine crystal. Rapid production of N2 and H2O within ∼10 ps is followed by delayed production of CO molecules beyond ns. We found that further decomposition towards the final products is inhibited by the formation of large metastable carbon- and oxygen-rich clusters with fractal geometry. In addition, we found distinct unimolecular and intermolecular reaction pathways, respectively, for the rapid N2 and H2O productions. read less
NOT USED (high confidence) R. Hernandez and A. Popov, “Molecular dynamics out of equilibrium: mechanics and measurables,” Wiley Interdisciplinary Reviews: Computational Molecular Science. 2014. link Times cited: 11
Abstract: Molecular dynamics is fundamentally the integration of the e… read more
Abstract: Molecular dynamics is fundamentally the integration of the equations of motion over a representation of an atomic and molecular system. The most rigorous choice for performing molecular dynamics entails the use of quantum‐mechanical equations of motion and a representation of the molecular system through all of its electrons and atoms. For most molecular problems involving at least hundreds of atoms, but generally many more, this is simply computationally prohibitive. Thus the art of molecular dynamics lies in choosing the representation and the appropriate equations of motion capable of addressing the requisite measurables. When used adroitly, it can provide both equilibrium (averaged) and time‐dependent properties of a molecular system. Many computational packages now exist that perform molecular dynamics simulations. They generally include force fields to represent the interactions between atoms and molecules (smoothing out electrons through the Born‐Oppenheimer approximation) and integrate the remaining particles classically. Despite these simplifications, all‐atom molecular dynamics remains computationally inaccessible if one includes the number of atoms required to simulate mesoscopic solvents. Here we use analytical models to demonstrate how molecular dynamics can be used to limit the solvent size in systems experiencing either equilibrium or nonequilibrium conditions. It is equally important to address the measurables (such as reaction rates) that are to be obtained prior to the generation of the data‐intensive trajectories. WIREs Comput Mol Sci 2014, 4:541–561. doi: 10.1002/wcms.1190 read less
NOT USED (high confidence) T. Zhou, L. Liu, W. Goddard, S. Zybin, and F. Huang, “ReaxFF reactive molecular dynamics on silicon pentaerythritol tetranitrate crystal validates the mechanism for the colossal sensitivity.,” Physical chemistry chemical physics : PCCP. 2014. link Times cited: 16
Abstract: Recently quantum mechanical (QM) calculations on a single Si… read more
Abstract: Recently quantum mechanical (QM) calculations on a single Si-PETN (silicon-pentaerythritol tetranitrate) molecule were used to explain its colossal sensitivity observed experimentally in terms of a unique Liu carbon-silyl nitro-ester rearrangement (R3Si-CH2-O-R2→ R3Si-O-CH2-R2). In this paper we expanded the study of Si-PETN from a single molecule to a bulk system by extending the ReaxFF reactive force field to describe similar Si-C-H-O-N systems with parameters optimized to reproduce QM results. The reaction mechanisms and kinetics of thermal decomposition of solid Si-PETN were investigated using ReaxFF reactive molecular dynamics (ReaxFF-RMD) simulations at various temperatures to explore the origin of the high sensitivity. We find that at lower temperatures, the decomposition of Si-PETN is initiated by the Liu carbon-silyl nitro-ester rearrangement forming Si-O bonds which is not observed in PETN. As the reaction proceeds, the exothermicity of Si-O bond formation promotes the onset of NO2 formation from N-OC bond cleavage which does not occur in PETN. At higher temperatures PETN starts to react by the usual mechanisms of NO2 dissociation and HONO elimination; however, Si-PETN remains far more reactive. These results validate the predictions from QM that the significantly increased sensitivity of Si-PETN arises from a unimolecular process involving the unusual Liu rearrangement but not from multi-molecular collisions. It is the very low energy barrier and the high exothermicity of the Si-O bond formation providing energy early in the decomposition process that is responsible. read less
NOT USED (high confidence) Q.-L. Yan, S. Zeman, P. E. S. Jimenez, T. Zhang, L. Pérez-Maqueda, and A. Elbeih, “The Mitigation Effect of Synthetic Polymers on Initiation Reactivity of CL-20: Physical Models and Chemical Pathways of Thermolysis,” Journal of Physical Chemistry C. 2014. link Times cited: 44
Abstract: In this paper, the thermal decomposition physical models of … read more
Abstract: In this paper, the thermal decomposition physical models of different CL-20 polymorph crystals and their polymer bonded explosives (PBXs) bonded by polymeric matrices using polyisobutylene (PIB), acrylonitrile butadiene rubber (NBR), styrene butadiene rubber (SBR), Viton A, and Fluorel binders are obtained and used to predict the temperature profiles of constant rate decomposition. The physical models are further supported by the detailed decomposition pathways simulated by a reactive molecular dynamics (ReaxFF-lg) code. It has been shown that both e-CL-20 and α-CL-20 decompose in the form of γ-CL-20, resulting in close activation energy (169 kJ mol–1) and physical model (first-order autoaccelerated model, AC1). Fluoropolymers could change the decomposition mechanism of e-CL-20 from the “first-order autocatalytic” model to a “three-dimensional nucleation and growth” model (A3), while the polymer matrices of Formex P1, Semtex, and C4 could change e-CL-20 decomposition from a single-step process to a multis... read less
NOT USED (high confidence) N. Ge et al., “Anisotropic responses and initial decomposition of condensed-phase β-HMX under shock loadings via molecular dynamics simulations in conjunction with multiscale shock technique.,” The journal of physical chemistry. B. 2014. link Times cited: 37
Abstract: Molecular dynamics simulations in conjunction with multiscal… read more
Abstract: Molecular dynamics simulations in conjunction with multiscale shock technique (MSST) are performed to study the initial chemical processes and the anisotropy of shock sensitivity of the condensed-phase HMX under shock loadings applied along the a, b, and c lattice vectors. A self-consistent charge density-functional tight-binding (SCC-DFTB) method was employed. Our results show that there is a difference between lattice vector a (or c) and lattice vector b in the response to a shock wave velocity of 11 km/s, which is investigated through reaction temperature and relative sliding rate between adjacent slipping planes. The response along lattice vectors a and c are similar to each other, whose reaction temperature is up to 7000 K, but quite different along lattice vector b, whose reaction temperature is only up to 4000 K. When compared with shock wave propagation along the lattice vectors a (18 Å/ps) and c (21 Å/ps), the relative sliding rate between adjacent slipping planes along lattice vector b is only 0.2 Å/ps. Thus, the small relative sliding rate between adjacent slipping planes results in the temperature and energy under shock loading increasing at a slower rate, which is the main reason leading to less sensitivity under shock wave compression along lattice vector b. In addition, the C-H bond dissociation is the primary pathway for HMX decomposition in early stages under high shock loading from various directions. Compared with the observation for shock velocities V(imp) = 10 and 11 km/s, the homolytic cleavage of N-NO2 bond was obviously suppressed with increasing pressure. read less
NOT USED (high confidence) N. Ge, Y. Wei, F. Zhao, X.-R. Chen, and G. Ji, “Pressure-induced metallization of condensed phase β-HMX under shock loadings via molecular dynamics simulations in conjunction with multi-scale shock technique,” Journal of Molecular Modeling. 2014. link Times cited: 15
NOT USED (high confidence) T. Zhou, H. Song, Y. Liu, and F. Huang, “Shock initiated thermal and chemical responses of HMX crystal from ReaxFF molecular dynamics simulation.,” Physical chemistry chemical physics : PCCP. 2014. link Times cited: 44
Abstract: To gain an atomistic-level understanding of the thermal and … read more
Abstract: To gain an atomistic-level understanding of the thermal and chemical responses of condensed energetic materials under thermal shock, we developed a thermal shock reactive dynamics (TS-RD) computational protocol using molecular dynamics simulation coupled with ReaxFF force field. β-Octahydro-1,3,5,7-tetranitro-1,3,5,7-tetrazocane (HMX) was selected as a a target explosive due to its wide usage in the military and industry. The results show that a thermal shock initiated by a large temperature gradient between the "hot" region and the "cold" region results in thermal expansion of the particles and induces a thermal-mechanical wave propagating back and forth in the system with an averaged velocity of 3.32 km s(-1). Heat propagating along the direction of thermal shock leads to a temperature increment of the system and thus chemical reaction initiation. Applying a continuum reactive heat conduction model combined with the temperature distribution obtained from the RD simulation, a heat conduction coefficient is derived as 0.80 W m(-1) K(-1). The chemical reaction mechanisms during thermal shock were analyzed, showing that the reaction is triggered by N-NO2 bond breaking followed by HONO elimination and ring fission. The propagation rates of the reaction front and reaction center are obtained to be 0.069 and 0.038 km s(-1), based on the time and spatial distribution of NO2. The pressure effect on the thermal shock was also investigated by employing uniaxial compression before the thermal shock. We find that compression significantly accelerates thermal-mechanical wave propagation and heat conduction, resulting in higher temperature and more excited molecules and thus earlier initiation and faster propagation of chemical reactions. read less
NOT USED (high confidence) O. Sergeev and A. Yanilkin, “Molecular dynamics simulation of combustion front propagation in a PETN single crystal,” Combustion, Explosion, and Shock Waves. 2014. link Times cited: 7
NOT USED (high confidence) O. Sergeev and A. Yanilkin, “Molecular dynamics simulation of combustion front propagation in a PETN single crystal,” Combustion, Explosion, and Shock Waves. 2014. link Times cited: 0
NOT USED (high confidence) E. Antillon, K. Banlusan, and A. Strachan, “Coarse grain model for coupled thermo-mechano-chemical processes and its application to pressure-induced endothermic chemical reactions,” Modelling and Simulation in Materials Science and Engineering. 2014. link Times cited: 14
Abstract: We extend a thermally accurate model for coarse grain dynami… read more
Abstract: We extend a thermally accurate model for coarse grain dynamics (Strachan and Holian 2005 Phys. Rev. Lett. 94 014301) to enable the description of stress-induced chemical reactions in the degrees of freedom internal to the mesoparticles. Similar to the breathing sphere model, we introduce an additional variable that describes the internal state of the particles and whose dynamics is governed both by an internal potential energy function and by interparticle forces. The equations of motion of these new variables are derived from a Hamiltonian and the model exhibits two desired features: total energy conservation and Galilean invariance. We use a simple model material with pairwise interactions between particles and study pressure-induced chemical reactions induced by hydrostatic and uniaxial compression. These examples demonstrate the ability of the model to capture non-trivial processes including the interplay between mechanical, thermal and chemical processes of interest in many applications. read less
NOT USED (high confidence) M. Fröhlich, T. Sewell, and D. Thompson, “Molecular dynamics simulations of shock waves in hydroxyl-terminated polybutadiene melts: mechanical and structural responses.,” The Journal of chemical physics. 2014. link Times cited: 22
Abstract: The mechanical and structural responses of hydroxyl-terminat… read more
Abstract: The mechanical and structural responses of hydroxyl-terminated cis-1,4-polybutadiene melts to shock waves were investigated by means of all-atom non-reactive molecular dynamics simulations. The simulations were performed using the OPLS-AA force field but with the standard 12-6 Lennard-Jones potential replaced by the Buckingham exponential-6 potential to better represent the interactions at high compression. Monodisperse systems containing 64, 128, and 256 backbone carbon atoms were studied. Supported shock waves were generated by impacting the samples onto stationary pistons at impact velocities of 1.0, 1.5, 2.0, and 2.5 km s(-1), yielding shock pressures between approximately 2.8 GPa and 12.5 GPa. Single-molecule structural properties (squared radii of gyration, asphericity parameters, and orientational order parameters) and mechanical properties (density, shock pressure, shock temperature, and shear stress) were analyzed using a geometric binning scheme to obtain spatio-temporal resolution in the reference frame centered on the shock front. Our results indicate that while shear stress behind the shock front is relieved on a ∼0.5 ps time scale, a shock-induced transition to a glass-like state occurs with a concomitant increase of structural relaxation times by several orders of magnitude. read less
NOT USED (high confidence) V. R. Soma, “Ultrashort laser pulse–matter interaction: Implications for high energy materials,” Pramana. 2014. link Times cited: 2
NOT USED (high confidence) L. He, T. Sewell, and D. Thompson, “Molecular dynamics simulations of shock waves in cis-1,4-polybutadiene melts,” Journal of Applied Physics. 2013. link Times cited: 14
Abstract: Molecular dynamics simulations of supported shock waves in m… read more
Abstract: Molecular dynamics simulations of supported shock waves in monodisperse melts of cis-1,4-polybutadiene initially at atmospheric pressure and T = 413 K were performed to study the shock-induced structural changes and post-shock relaxation. Simulations were performed for Rankine-Hugoniot shock pressures between 7.22 GPa and 8.26 GPa using the united-atom force field due to Smith and Paul [G. D. Smith and W. Paul, J. Phys. Chem. A 102, 1200 (1998)] for systems composed of chains containing 32, 64, or 128 united atoms. The sensitivity of the results to the non-bonded interaction potential was studied by comparing results obtained using the Lennard-Jones 12–6 potential from the original Smith and Paul force field to ones obtained when the 12–6 potential was replaced by the Buckingham exponential–6 potential. Several structural and mechanical properties were studied as functions of distance (time) behind the shock front. Bulk relaxation was characterized by calculating profiles of temperature, density, and prin... read less
NOT USED (high confidence) J. Huang and M. Meuwly, “Force Field Treatment of Proton and Hydrogen Transfer in Molecular Systems.” 2013. link Times cited: 1
Abstract: Proton and hydrogen-bonded motifs are amongst the most widel… read more
Abstract: Proton and hydrogen-bonded motifs are amongst the most widely occurring patterns in chemistry and biology. Besides their intrinsic chemical significance, they also display a fascinating range of dynamical effects including highly quantum behavior or strong coupling between inter and intramolecular degrees of freedom. In chemical and biological systems, rates for proton transfer (PT) or hydrogen transfer (HT) can cover the picosecond to millisecond range, which corresponds to a few kilocalories per mole up to 20 kcal mol−1. From a computational perspective, this implies that the energetics along a specific motif has to be extensively sampled in order to converge the experimental observables. Possible approaches to treat the energetics include ab initio molecular dynamics (AIMD) simulations, mixed quantum mechanical/molecular mechanics (QM/MM) calculations, and more or less empirical parameterizations of the intermolecular interactions based on model potentials or parameterized fits to rigorous quantum chemical calculations. In this chapter, we discuss QM/MM embedding schemes into an empirical force field based on fitting high-quality (Moller–Plesset perturbation theory (MP2) with a large basis set) quantum chemical calculations, which allows the explicit treatment of the long-time dynamics. The essential feature of this procedure is its accuracy, flexibility, and suitability for either QM or classical treatments of the nuclear dynamics. The present chapter discusses the theory, implementation, and applications of this embedding scheme and discusses it with regard to other existing ways to treat PT or HT in strongly and weakly coupled systems. PT and HT reactions are fundamental in chemistry and biology. Although the famous Grotthuss shuttling mechanism [1] was introduced more than 200 years ago, notable progress in understanding atomistic details underlying the process has only been achieved recently with modern spectroscopic techniques and high-performance computer simulations [2]. From an experimental point of view, infrared (IR) studies [3–5] were successful in probing the vibrational dynamics of hydrogen bonds. However, a complete assignment/interpretation of the spectra read less
NOT USED (high confidence) A. Bochevarov et al., “Jaguar: A high-performance quantum chemistry software program with strengths in life and materials sciences,” International Journal of Quantum Chemistry. 2013. link Times cited: 1287
Abstract: Jaguar is an ab initio quantum chemical program that special… read more
NOT USED (high confidence) S. Root et al., “Shock compression of hydrocarbon foam to 200 GPa: Experiments, atomistic simulations, and mesoscale hydrodynamic modeling,” Journal of Applied Physics. 2013. link Times cited: 28
Abstract: Hydrocarbon foams are versatile materials extensively used i… read more
Abstract: Hydrocarbon foams are versatile materials extensively used in high energy-density physics (HEDP) experiments. However, little data exist above 100 GPa, where knowledge of the behavior is particularly important for designing, analyzing, and optimizing HEDP experiments. The complex internal structure and properties of foam call for a multi-scale modeling effort validated by experimental data. We present results from experiments, classical molecular dynamics simulations, and mesoscale hydrodynamic modeling of poly(4-methyl-1-pentene) (PMP) foams under strong shock compression. Experiments conducted using the Z-machine at Sandia National Laboratories shock compress ∼0.300 g/cm3 density PMP foams to 185 GPa. Molecular dynamics (MD) simulations model shock compressed PMP foam and elucidate behavior of the heterogeneous foams at high pressures. The MD results show quantitative agreement with the experimental data, while providing additional information about local temperature and dissociation. Three-dimensional ... read less
NOT USED (high confidence) M. Fröhlich and T. Sewell, “Pivot Algorithm and Push‐off Method for Efficient System Generation of All‐Atom Polymer Melts: Application to Hydroxyl‐Terminated Polybutadiene,” Macromolecular Theory and Simulations. 2013. link Times cited: 7
Abstract: Procedures used to generate initial conditions for all-atom … read more
Abstract: Procedures used to generate initial conditions for all-atom molecular dynamics (MD) simulations of amorphous polymer systems are described. The pivot algorithm is applied directly to produce unbranched hydroxyl-terminated cis-1,4-polybutadiene molecules with chain lengths ranging from 8 to 64 monomeric units (32 to 256 carbon atoms), based on the OPLS all-atom force field. The generated molecular configurations are characteristic of (pseudo-) ideal conditions and therefore exhibit the same properties as polymer chains in a melt. We analyze both the mean squared chain dimensions and their distributions in order to validate the correctness of this approach. The chains are efficiently packed into 3d-periodic simulation cells using the push-off method and subsequently equilibrated by standard MD. Together, the pivot algorithm and push-off method provide the means for efficient generation of equilibrated dense polymer melts for multi-million-atom systems. read less
NOT USED (high confidence) W. Song and S.-jin Zhao, “Development of the ReaxFF reactive force field for aluminum–molybdenum alloy,” Journal of Materials Research. 2013. link Times cited: 10
Abstract: We have developed a reactive force field within the ReaxFF f… read more
Abstract: We have developed a reactive force field within the ReaxFF framework to accurately describe reactions involving aluminum–molybdenum alloy, which are part parameters of Al–O–Mo ternary system metastable intermolecular composites. The parameters are optimized from a training set, whose data come from density functional theory (DFT) calculations and experimental value, such as heat of formation, geometry data, and equation of states, which are reproduced well by ReaxFF. Body-centered cubic molybdenum’s surface energy, vacancy formation, and two transformational paths, Bain and trigonal paths are calculated to validate the ReaxFF ability describing the defects and deformations. Some structures’ elastic constant and phonon are calculated by DFT and ReaxFF to predict the structures’ mechanics and kinetic stability. All those results indicate that the fitted parameters can describe the energy difference of various structures under various circumstances and generally represent the diffusion property but cannot reproduce the elasticity and phonon spectra so well. read less
NOT USED (high confidence) N. Goldman, S. Srinivasan, S. Hamel, L. Fried, M. Gaus, and M. Elstner, “Determination of a Density Functional Tight Binding Model with an Extended Basis Set and Three-Body Repulsion for Carbon Under Extreme Pressures and Temperatures,” Journal of Physical Chemistry C. 2013. link Times cited: 24
Abstract: We report here on development of a density functional tight … read more
Abstract: We report here on development of a density functional tight binding (DFTB) simulation approach for carbon under extreme pressures and temperatures that includes an expanded basis set and an environmentally dependent repulsive energy. We find that including d-orbital interactions in the DFTB Hamiltonian improves determination of the electronic states at high pressure–temperature conditions, compared to standard DFTB implementations that utilize s- and p-orbitals only for carbon. We then determine a three-body repulsive energy through fitting to diamond, BC8, and simple cubic cold compression curve data, as well pressures from metallic liquid configurations from density functional theory (DFT) simulations. Our new model (DFTB-p3b) yields approximately 2 orders of magnitude increase in computational efficiency over standard DFT while retaining its accuracy for condensed phases of carbon under a wide range of conditions, including the metallic liquid phase at conditions up to 2000 GPa and 30 000 K. Our result... read less
NOT USED (high confidence) S. Naserifar, L. Liu, W. Goddard, T. Tsotsis, and M. Sahimi, “Toward a Process-Based Molecular Model of SiC Membranes. 1. Development of a Reactive Force Field,” Journal of Physical Chemistry C. 2013. link Times cited: 38
Abstract: A broad class of important materials, such as carbon molecul… read more
Abstract: A broad class of important materials, such as carbon molecular sieves, silicon carbide (SiC), and silicon nitride, are fabricated by temperature-controlled pyrolysis of preceramic polymers. In particular, the fabrication of SiC membranes by pyrolysis of a polymer precursor that contains Si is quite attractive for separation of hydrogen from other gases. It has been quite difficult to extract atomistic-scale information about such SiC membranes since they are amorphous. In principle, ab initio quantum mechanics (QM) can provide information about the structure of the amorphous systems. However, to determine the structure of the SiC membrane layer one should capture in the simulations the various reactive processes involved in forming the layer. This requires QM simulations on systems with about 3000 atoms per cell at temperature of 1200 K for microseconds, which are far beyond the current QM capabilities. Instead, this paper extends the ReaxFF reactive force field, validated for high temperature reactions of other materials, to describe the processes involved in the thermal decomposition of hydridopolycarbosilane (HPCS) to form SiC nanoporous membranes. First, we carry out QM calculations on models meant to capture important reaction steps and structures. Then, we develop a model of the HPCS polymer and utilize ReaxFF to describe the thermal degradation and decomposition of the polymer as the system is heated in the molecular dynamics (MD) simulations. Analysis of the pyrolysis studies and their results leads to various quantities that can be compared with experimental data. Good agreement is found between the data and the results of the MD simulations. read less
NOT USED (high confidence) T.-R. Shan, R. Wixom, A. Mattsson, and A. Thompson, “Atomistic simulation of orientation dependence in shock-induced initiation of pentaerythritol tetranitrate.,” The journal of physical chemistry. B. 2013. link Times cited: 53
Abstract: The dependence of the reaction initiation mechanism of penta… read more
Abstract: The dependence of the reaction initiation mechanism of pentaerythritol tetranitrate (PETN) on shock orientation and shock strength is investigated with molecular dynamics simulations using a reactive force field and the multiscale shock technique. In the simulations, a single crystal of PETN is shocked along the [110], [001], and [100] orientations with shock velocities in the range 3-10 km/s. Reactions occur with shock velocities of 6 km/s or stronger, and reactions initiate through the dissociation of nitro and nitrate groups from the PETN molecules. The most sensitive orientation is [110], while [100] is the most insensitive. For the [001] orientation, PETN decomposition via nitro group dissociation is the dominant reaction initiation mechanism, while for the [110] and [100] orientations the decomposition is via mixed nitro and nitrate group dissociation. For shock along the [001] orientation, we find that CO-NO(2) bonds initially acquire more kinetic energy, facilitating nitro dissociation. For the other two orientations, C-ONO(2) bonds acquire more kinetic energy, facilitating nitrate group dissociation. read less
NOT USED (high confidence) N. Ge, Y. Wei, G. Ji, X.-R. Chen, F. Zhao, and D. Wei, “Initial decomposition of the condensed-phase β-HMX under shock waves: molecular dynamics simulations.,” The journal of physical chemistry. B. 2012. link Times cited: 58
Abstract: We have performed quantum-based multiscale simulations to st… read more
Abstract: We have performed quantum-based multiscale simulations to study the initial chemical processes of condensed-phase octahydro-1,3,5,7-tetranitro-1,3,5,7-tetrazocine (HMX) under shock wave loading. A self-consistent charge density-functional tight-binding (SCC-DFTB) method was employed. The results show that the initial decomposition of shocked HMX is triggered by the N-NO(2) bond breaking under the low velocity impact (8 km/s). As the shock velocity increases (11 km/s), the homolytic cleavage of the N-NO(2) bond is suppressed under high pressure, the C-H bond dissociation becomes the primary pathway for HMX decomposition in its early stages. It is accompanied by a five-membered ring formation and hydrogen transfer from the CH(2) group to the -NO(2) group. Our simulations suggest that the initial chemical processes of shocked HMX are dependent on the impact velocity, which gain new insights into the initial decomposition mechanism of HMX upon shock loading at the atomistic level, and have important implications for understanding and development of energetic materials. read less
NOT USED (high confidence) T. Zhou, S. Zybin, Y. Liu, F. Huang, and W. Goddard, “Anisotropic shock sensitivity for β-octahydro-1,3,5,7-tetranitro-1,3,5,7-tetrazocine energetic material under compressive-shear loading from ReaxFF-lg reactive dynamics simulations,” Journal of Applied Physics. 2012. link Times cited: 75
Abstract: We report here the predictions on anisotropy of shock sensit… read more
Abstract: We report here the predictions on anisotropy of shock sensitivity and of chemical process initiation in single crystal β-octahydro-1,3,5,7-tetranitro-1,3,5,7-tetrazocine (β-HMX) using compressive shear reactive dynamics (CS-RD) model with ReaxFF-lg reactive force field. Analysis of resolved shear stress induced by uniaxial compression along three shock directions normal to (110), (011), and (010) planes leads to identify eight slip systems as candidates for shear deformation. For each of the eight slip systems, non-equilibrium reactive dynamics simulations were carried out to determine thermal, mechanical, and chemical responses to shear deformation. Shock direction normal to (010) plane exhibits large shear stress barriers arising from steric hindrance between molecules of adjacent layers leading to local dramatic energy and temperature increases under shear flow that in turn accelerate chemical bond breaking and initial product formation processes, promoting further molecular decomposition and eventually transition to detonation. This suggests that single crystal β-HMX is sensitive to shocks in direction normal to (010) plane. Shock directions normal to (110) and (011) planes reveal significantly less steric hindrance, leading to more modest energy and temperature increases followed by slower chemical reaction initiation. Thus, shock directions normal to (110) and (011) planes are less sensitive than shock direction normal to (010) plane, which agree with interpretations from currently available plate impact experiments on HMX. This validation of CS-RD and ReaxFF for characterizing sensitivity of single crystal energetic materials indicates that these methods can be applied to study sensitivity for more complex polymer bonded explosives and solid composite propellants having complex microstructures, corrugated interfaces, as well as defects. read less
NOT USED (high confidence) C. Zhang, Y. Ma, and D. Jiang, “Charge transfer in TATB and HMX under extreme conditions,” Journal of Molecular Modeling. 2012. link Times cited: 14
NOT USED (high confidence) P.-A. Cazade, J. Huang, J. Yosa, J. J. Szymczak, and M. Meuwly, “Atomistic simulations of reactive processes in the gas- and condensed-phase,” International Reviews in Physical Chemistry. 2012. link Times cited: 4
Abstract: This review focuses on force-field-based approaches to inves… read more
Abstract: This review focuses on force-field-based approaches to investigate – through computer simulations – reactive processes in chemical and biological systems. Both, reactions in the gas-phase and in condensed-phase environments are discussed and opportunities and the potential for further developments are pointed out. Where available, results are compared with alternative methods and the advantages and drawbacks of the methods are compared. Particular applications include vibrationally and electronically induced (photo)dissociation of small molecules, proton transfer in the gas- and condensed phase and ligand un- and re-binding in proteins. read less
NOT USED (high confidence) F. Guo, X. Cheng, and H. Zhang, “Reactive molecular dynamics simulation of solid nitromethane impact on (010) surfaces induced and nonimpact thermal decomposition.,” The journal of physical chemistry. A. 2012. link Times cited: 54
Abstract: Which is the first step in the decomposition process of nitr… read more
Abstract: Which is the first step in the decomposition process of nitromethane is a controversial issue, proton dissociation or C-N bond scission. We applied reactive force field (ReaxFF) molecular dynamics to probe the initial decomposition mechanisms of nitromethane. By comparing the impact on (010) surfaces and without impact (only heating) for nitromethane simulations, we found that proton dissociation is the first step of the pyrolysis of nitromethane, and the C-N bond decomposes in the same time scale as in impact simulations, but in the nonimpact simulation, C-N bond dissociation takes place at a later time. At the end of these simulations, a large number of clusters are formed. By analyzing the trajectories, we discussed the role of the hydrogen bond in the initial process of nitromethane decompositions, the intermediates observed in the early time of the simulations, and the formation of clusters that consisted of C-N-C-N chain/ring structures. read less
NOT USED (high confidence) P. Pahari and S. Chaturvedi, “Determination of best-fit potential parameters for a reactive force field using a genetic algorithm,” Journal of Molecular Modeling. 2012. link Times cited: 32
NOT USED (high confidence) B. Chen et al., “ReaxFF Reactive Force Field for Molecular Dynamics Simulations of Lignite Depolymerization in Supercritical Methanol with Lignite-Related Model Compounds,” Energy & Fuels. 2012. link Times cited: 34
Abstract: To investigate the detailed mechanisms for lignite methanoly… read more
Abstract: To investigate the detailed mechanisms for lignite methanolysis, we used ReaxFF reactive force field to perform a series of molecular dynamics simulations (MDSs) on a unimolecular model compound. The α-O-4 and β-O-4 types of lignite-related model compounds were selected as representatives of linkages in lignites. The reaction products predicted by ReaxFF MDSs are consistent with those from experimental results reported. The initiation reaction observed in ReaxFF MDSs involving the ether linkage cleavage and methanol participation closely matches the results observed from previously reported experiments. The agreement of these results with available experimental observations demonstrates that ReaxFF MDSs can give an atomistic description of the initiation mechanism for methanolysis and provide useful insights into the complicated reaction processes. read less
NOT USED (high confidence) L. He, T. Sewell, and D. Thompson, “Molecular dynamics simulations of shock waves in oriented nitromethane single crystals: plane-specific effects.,” The Journal of chemical physics. 2012. link Times cited: 10
Abstract: Molecular dynamics simulations of supported shock waves (sho… read more
Abstract: Molecular dynamics simulations of supported shock waves (shock pressure P(s) ∼ 15 GPa) propagating along the [110], [011], [101], and [111] directions in crystalline nitromethane initially at T = 200 K were performed using the nonreactive Sorescu-Rice-Thompson force field [D. C. Sorescu, B. M. Rice, and D. L. Thompson, J. Phys. Chem. B 104, 8406 (2000)]. These simulations, combined with those from a preceding study of shocks propagating along [100], [010], and [001] directions in nitromethane for similar conditions of temperature and shock pressure [L. He, T. D. Sewell, and D. L. Thompson, J. Chem. Phys. 134, 124506 (2011)], have been used to study the post-shock relaxation phenomena. Shocks along [010] and [101] lead to a crystal-crystal structure transformation. Shocks propagating along [011], [110], [111], [100], and [001] exhibit plane-specific disordering, which was characterized by calculating as functions of time the 1D mean square displacement (MSD), 2D radial distribution function (RDF), and 2D orientation order parameter P(2)(θ) in orthogonal planes mutually perpendicular to the shock plane; and by calculating as functions of distance behind the shock front the Cartesian components of intermolecular, intramolecular, and total kinetic energies. The 2D RDF results show that the structural disordering for shocks along [100], [110], and [111] is strongly plane-specific; whereas for shocks along [001] and [011], the loss of crystal structural order is almost equivalent in the orthogonal planes perpendicular to the shock plane. Based on the entire set of simulations, there is a trend for the most extensive disordering to occur in the (010) and (110) planes, less extensive disordering to occur in the (100) plane, and essentially no disordering to occur in the (001) plane. The 2D P(2)(θ) and 1D MSD profiles show, respectively, that the orientational and translational disordering is plane-specific, which results in the plane-specific structural disordering observed in the 2D RDF. By contrast, the kinetic energy partitioning and redistribution do not exhibit plane specificity, as shown by the similarity of spatial profiles of the Cartesian components of the intermolecular, intramolecular, and total kinetic energies in orthogonal planes perpendicular to the shock plane. read less
NOT USED (high confidence) Q. An, S. Zybin, W. Goddard, A. Jaramillo-Botero, M. Blanco, and S. Luo, “Elucidation of the Dynamics for Hot-Spot Initiation at Nonuniform Interfaces of Highly Shocked Materials,” Physical Review B. 2011. link Times cited: 83
Abstract: The fundamental processes in shock-induced instabilities of … read more
Abstract: The fundamental processes in shock-induced instabilities of materials remain obscure, particularly for detonation of energetic materials. We simulated these processes at the atomic scale on a realistic model of a polymer-bonded explosive (3,695,375 atoms/cell) and observed that a hot spot forms at the nonuniform interface, arising from shear relaxation that results in shear along the interface that leads to a large temperature increase that persists long after the shock front has passed the interface. For energetic materials this temperature increase is coupled to chemical reactions that lead to detonation. We show that decreasing the density of the binder eliminates the hot spot. read less
NOT USED (high confidence) L. Liu, Y. Liu, S. Zybin, H. Sun, and W. Goddard, “ReaxFF-lg: correction of the ReaxFF reactive force field for London dispersion, with applications to the equations of state for energetic materials.,” The journal of physical chemistry. A. 2011. link Times cited: 374
Abstract: The practical levels of density functional theory (DFT) for … read more
Abstract: The practical levels of density functional theory (DFT) for solids (LDA, PBE, PW91, B3LYP) are well-known not to account adequately for the London dispersion (van der Waals attraction) so important in molecular solids, leading to equilibrium volumes for molecular crystals ~10-15% too high. The ReaxFF reactive force field is based on fitting such DFT calculations and suffers from the same problem. In the paper we extend ReaxFF by adding a London dispersion term with a form such that it has low gradients (lg) at valence distances leaving the already optimized valence interactions intact but behaves as 1/R(6) for large distances. We derive here these lg corrections to ReaxFF based on the experimental crystal structure data for graphite, polyethylene (PE), carbon dioxide, and nitrogen and for energetic materials: hexahydro-1,3,5-trinitro-1,3,5-s-triazine (RDX), pentaerythritol tetranitrate (PETN), 1,3,5-triamino-2,4,6-trinitrobenzene (TATB), and nitromethane (NM). After this dispersion correction the average error of predicted equilibrium volumes decreases from 18.5 to 4.2% for the above systems. We find that the calculated crystal structures and equation of state with ReaxFF-lg are in good agreement with experimental results. In particular, we examined the phase transition between α-RDX and γ-RDX, finding that ReaxFF-lg leads to excellent agreement for both the pressure and volume of this transition occurring at ~4.8 GPa and ~2.18 g/cm(3) density from ReaxFF-lg vs 3.9 GPa and ~2.21 g/cm(3) from experiment. We expect ReaxFF-lg to improve the descriptions of the phase diagrams for other energetic materials. read less
NOT USED (high confidence) I. Schweigert and B. Dunlap, “Shattering dissociation in high-energy molecular collisions between nitrate esters.,” The Journal of chemical physics. 2011. link Times cited: 0
Abstract: We present ab initio molecular dynamics simulations of head-… read more
Abstract: We present ab initio molecular dynamics simulations of head-on collisions between ethyl nitrate molecules at collisional energies from 200 to 1200 kJ/mol. Above a threshold energy, an increasing fraction of the collisions led to rapid dissociation on impact--"shattering." The probability of the shattering dissociation was derived from the quasiclassical trajectories sampling the initial vibrational motion at T(vib) = 300 K. Even for the zero impact parameter and a fixed orientation considered, the observed dissociation probability exhibited a wide spread (much larger than kT(vib)) as a function of the collision energy. This is attributed to variations in the initial vibrational phase. We propose a closed-form expression for the energy-dependent dissociation probability that captures the dependence on the phase and use it to analyze the probability of the shattering dissociation of a larger nitrate ester, pentaerythritol tetranitrate. read less
NOT USED (high confidence) T. Watanabe, “Dynamic bond-order force field,” Journal of Computational Electronics. 2011. link Times cited: 8
NOT USED (high confidence) L. Liu, C. Bai, H. Sun, and W. Goddard, “Mechanism and kinetics for the initial steps of pyrolysis and combustion of 1,6-dicyclopropane-2,4-hexyne from ReaxFF reactive dynamics.,” The journal of physical chemistry. A. 2011. link Times cited: 99
Abstract: We report the kinetic analysis and mechanism for the initial… read more
Abstract: We report the kinetic analysis and mechanism for the initial steps of pyrolysis and combustion of a new fuel material, 1,6-dicyclopropane-2,4-hexyne, that has enormous heats of pyrolysis and combustion, making it a potential high-energy fuel or fuel additive. These studies employ the ReaxFF force field for reactive dynamics (RD) simulations of both pyrolysis and combustion processes for both unimolecular and multimolecular systems. We find that both pyrolysis and combustion initiate from unimolecular reactions, with entropy-driven reactions being most important in both processes. Pyrolysis initiates with extrusion of an ethylene molecule from the fuel molecule and is followed quickly by isomerization of the fuel molecule, which induces additional radicals that accelerate the pyrolysis process. In the combustion process, we find three distinct mechanisms for the O(2) attack on the fuel molecule: (1) attack on the cyclopropane, ring expanding to form the cyclic peroxide which then decomposes; (2) attack onto the central single bond of the diyne which then fissions to form two C(5)H(5)O radicals; (3) attack on the alkyne-cyclopropane moiety to form a seven-membered ring peroxide which then decomposes. Each of these unimolecular combustion processes releases energy that induces additional radicals to accelerate the combustion process. Here oxygen has major effects both as the radical acceptor and as the radical producer. We extract both the effective activation energy and the effective pre-exponential factor by kinetic analysis of pyrolysis and combustion from these ReaxFF simulations. The low value of the derived effective activation energy (26.18 kcal/mol for pyrolysis and 16.40 kcal/mol for combustion) reveals the high activity of this fuel molecule. read less
NOT USED (high confidence) S.-ping Han, A. V. van Duin, W. Goddard, and A. Strachan, “Thermal decomposition of condensed-phase nitromethane from molecular dynamics from ReaxFF reactive dynamics.,” The journal of physical chemistry. B. 2011. link Times cited: 98
Abstract: We studied the thermal decomposition and subsequent reaction… read more
Abstract: We studied the thermal decomposition and subsequent reaction of the energetic material nitromethane (CH(3)NO(2)) using molecular dynamics with ReaxFF, a first principles-based reactive force field. We characterize the chemistry of liquid and solid nitromethane at high temperatures (2000-3000 K) and density 1.97 g/cm(3) for times up to 200 ps. At T = 3000 K the first reaction in the decomposition of nitromethane is an intermolecular proton transfer leading to CH(3)NOOH and CH(2)NO(2). For lower temperatures (T = 2500 and 2000 K) the first reaction during decomposition is often an isomerization reaction involving the scission of the C-N bond the formation of a C-O bond to form methyl nitrate (CH(3)ONO). Also at very early times we observe intramolecular proton transfer events. The main product of these reactions is H(2)O which starts forming following those initiation steps. The appearance of H(2)O marks the beginning of the exothermic chemistry. Recent quantum-mechanics-based molecular dynamics simulations on the chemical reactions and time scales for decomposition of a crystalline sample heated to T = 3000 K for a few picoseconds are in excellent agreement with our results, providing an important, direct validation of ReaxFF. read less
NOT USED (high confidence) D. Dlott, “New developments in the physical chemistry of shock compression.,” Annual review of physical chemistry. 2011. link Times cited: 69
Abstract: This review discusses new developments in shock compression … read more
Abstract: This review discusses new developments in shock compression science with a focus on molecular media. Some basic features of shock and detonation waves, nonlinear excitations that can produce extreme states of high temperature and high pressure, are described. Methods of generating and detecting shock waves are reviewed, especially those using tabletop lasers that can be interfaced with advanced molecular diagnostics. Newer compression methods such as shockless compression and precompression shock that generate states of cold dense molecular matter are discussed. Shock compression creates a metallic form of hydrogen, melts diamond, and makes water a superionic liquid with unique catalytic properties. Our understanding of detonations at the molecular level has improved a great deal as a result of advanced nonequilibrium molecular simulations. Experimental measurements of detailed molecular behavior behind a detonation front might be available soon using femtosecond lasers to produce nanoscale simulated detonation fronts. read less
NOT USED (high confidence) L. He, T. Sewell, and D. Thompson, “Molecular dynamics simulations of shock waves in oriented nitromethane single crystals.,” The Journal of chemical physics. 2011. link Times cited: 32
Abstract: The structural relaxation of crystalline nitromethane initia… read more
Abstract: The structural relaxation of crystalline nitromethane initially at T = 200 K subjected to moderate (~15 GPa) supported shocks on the (100), (010), and (001) crystal planes has been studied using microcanonical molecular dynamics with the nonreactive Sorescu-Rice-Thompson force field [D. C. Sorescu, B. M. Rice, and D. L. Thompson, J. Phys. Chem. B 104, 8406 (2000)]. The responses to the shocks were determined by monitoring the mass density, the intermolecular, intramolecular, and total temperatures (average kinetic energies), the partitioning of total kinetic energy among Cartesian directions, the radial distribution functions for directions perpendicular to those of shock propagation, the mean-square displacements in directions perpendicular to those of shock propagation, and the time dependence of molecular rotational relaxation as a function of time. The results show that the mechanical response of crystalline nitromethane strongly depends on the orientation of the shock wave. Shocks propagating along [100] and [001] result in translational disordering in some crystal planes but not in others, a phenomenon that we refer to as plane-specific disordering; whereas for [010] the shock-induced stresses are relieved by a complicated structural rearrangement that leads to a paracrystalline structure. The plane-specific translational disordering is more complete by the end of the simulations (~6 ps) for shock propagation along [001] than along [100]. Transient excitation of the intermolecular degrees of freedom occurs in the immediate vicinity of the shock front for all three orientations; the effect is most pronounced for the [010] shock. In all three cases excitation of molecular vibrations occurs more slowly than the intermolecular excitation. The intermolecular and intramolecular temperatures are nearly equal by the end of the simulations, with 400-500 K of net shock heating. Results for two-dimensional mean-square molecular center-of-mass displacements, calculated as a function of time since shock wave passage in planes perpendicular to the direction of shock propagation, show that the molecular translational mobility in the picoseconds following shock wave passage is greatest for [001] and least for the [010] case. In all cases the root-mean-square center-of-mass displacement is small compared to the molecular diameter of nitromethane on the time scale of the simulations. The calculated time scales for the approach to thermal equilibrium are generally consistent with the predictions of a recent theoretical analysis due to Hooper [J. Chem. Phys. 132, 014507 (2010)]. read less
NOT USED (high confidence) A. Jaramillo-Botero et al., “First-principles-based multiscale, multiparadigm molecular mechanics and dynamics methods for describing complex chemical processes.,” Topics in current chemistry. 2011. link Times cited: 15
NOT USED (high confidence) S. Zybin, W. Goddard, P. Xu, A. Duin, and A. Thompson, “Physical mechanism of anisotropic sensitivity in pentaerythritol tetranitrate from compressive-shear reaction dynamics simulations,” Applied Physics Letters. 2010. link Times cited: 93
Abstract: We propose computational protocol (compressive shear reactiv… read more
Abstract: We propose computational protocol (compressive shear reactive dynamics) utilizing the ReaxFF reactive force field to study chemical initiation under combined shear and compressive load. We apply it to predict the anisotropic initiation sensitivity observed experimentally for shocked pentaerythritol tetranitrate single crystals. For crystal directions known to be sensitive we find large stress overshoots and fast temperature increase that result in early bond-breaking processes whereas insensitive directions exhibit small stress overshoot, lower temperature increase, and little bond dissociation. These simulations confirm the model of steric hindrance to shear and capture the thermochemical processes dominating the phenomena of shear-induced chemical initiation. read less
NOT USED (high confidence) R. Dawes, A. Siavosh-Haghighi, T. Sewell, and D. Thompson, “Shock-induced melting of (100)-oriented nitromethane: Energy partitioning and vibrational mode heating.,” The Journal of chemical physics. 2009. link Times cited: 24
Abstract: A study of the structural relaxation of nitromethane subsequ… read more
Abstract: A study of the structural relaxation of nitromethane subsequent to shock loading normal to the (100) crystal plane performed using molecular dynamics and a nonreactive potential was reported recently [J. Chem. Phys. 131, 064503 (2009)]. Starting from initial temperatures of T(0)=50 and 200 K, shocks were simulated using impact velocities U(p) ranging from 0.5 to 3.0 km s(-1); clear evidence of melting was obtained for shocks initiated with impacts of 2.0 km s(-1) and higher. Here, we report the results of analyses of those simulation data using a method based on the Eckart frame normal-mode analysis that allows partitioning of the kinetic energy among the molecular degrees of freedom. A description of the energy transfer is obtained in terms of average translational and rotational kinetic energies in addition to the rates of individual vibrational mode heating. Generally, at early times postshock a large superheating of the translational and rotational degrees of freedom (corresponding to phonon modes of the crystal) is observed. The lowest frequency vibrations (gateway modes) are rapidly excited and also exhibit superheating. Excitation of the remaining vibrational modes occurs more slowly. A rapid, early excitation of the symmetric C-H stretch mode was observed for the shock conditions T(0)=50 K and U(p)=2.0 km s(-1) due to a combination of favorable alignment of molecular orientation with the shock direction and frequency matching between the vibration and shock velocity. read less
NOT USED (high confidence) E. Salmon, A. Duin, F. Lorant, P. Marquaire, and W. Goddard, “Early maturation processes in coal. Part 2: Reactive dynamics simulations using the ReaxFF reactive force field on Morwell Brown coal structures,” Organic Geochemistry. 2009. link Times cited: 176
NOT USED (high confidence) C. Zhou, J. Wu, L. Chen, Y. Wang, H. Cheng, and R. C. Forrey, “Force field for copper clusters and nanoparticles,” Journal of Computational Chemistry. 2009. link Times cited: 2
Abstract: An atomic force field for simulating copper clusters and nan… read more
NOT USED (high confidence) B. Szyja, A. Jansen, T. Verstraelen, and R. V. van Santen, “Molecular dynamics study of the silica-water-SDA interactions.,” Physical chemistry chemical physics : PCCP. 2009. link Times cited: 15
Abstract: In this paper we have applied the molecular dynamics simulat… read more
Abstract: In this paper we have applied the molecular dynamics simulations in order to analyse the role of the structure directing tetrapropylammonium ions in the aggregation process that leads to silicalite formation. We address the specific question of how the interactions between silica precursor species and tetrapropylammonium ions/water evolve during the formation of the larger aggregates, that show initial micropore formation from more elementary building blocks. We have followed the dynamics and changes in the position of the tetrapropylammonium ions into the formation of TPA-Si22 complexes. Moreover, the analysis based on the geometries of the systems being studied as well as the radial distribution function allowed us to predict the location of the TPA cations in fully formed nanoslabs. An interesting result is reported that the template cannot be accommodated any more in the newly formed cavities, but is pushed out of the channel like cavities to positions where in a later stage channel cross sections can be formed. read less
NOT USED (high confidence) A. Siavosh-Haghighi, R. Dawes, T. Sewell, and D. Thompson, “Shock-induced melting of (100)-oriented nitromethane: structural relaxation.,” The Journal of chemical physics. 2009. link Times cited: 22
Abstract: Molecules subjected to shock waves will, in general, undergo… read more
Abstract: Molecules subjected to shock waves will, in general, undergo significant intramolecular distortion and exhibit large amplitude orientational and translational displacements relative to the unshocked material. The analysis of molecular dynamics simulations of strongly perturbed materials is complicated, particularly when the goal is to express time-dependent molecular-scale properties in terms of structural or geometric descriptors/properties defined for molecules in the equilibrium geometry. We illustrate the use of the Eckart-Sayvetz condition in a molecular dynamics study of the response of crystalline nitromethane subjected to supported shock waves propagating normal to (100). The simulations were performed with the nonreactive but vibrationally accurate force field due to Sorescu et al. [J. Phys. Chem. B 104, 8406 (2000)]. Shocks were initiated with impact velocities of U(p)=0.5, 1.0, 2.0, and 3.0 km s(-1) in crystals at initial temperatures of T0=50 and 200 K. Statistical precision in the analysis was enhanced through the use of a spatiotemporal reference frame centered on the advancing shock front, which was located as a function of time using the gradient of the kinetic energy along the shock direction. The Eckart-Sayvetz condition provides a rigorous approach by which the alignment can be obtained between a coordinate frame for a perturbed molecule and one in a convenient reference frame (e.g., one based on the equilibrium crystal structure) for analyses of the molecules in the material as the system evolves toward equilibrium. Structural and dynamic properties of the material corresponding to orientation in the lattice, translational symmetry, and mass transport (orientational order parameters, two dimensional radial distribution functions, and self-diffusion coefficients, respectively) were computed as functions of time with 4 fs resolution. The results provide clear evidence of melting for shocks initiated by impacts of at least U(p)=2.0 km s(-1) and provide insights into the evolution of changes at the molecular-mode level associated with the onset of the melting instability in shocked crystal. read less
NOT USED (high confidence) J. Ojwang, R. A. Santen, G. Kramer, A. Duin, and W. Goddard, “Parametrization of a reactive force field for aluminum hydride.,” The Journal of chemical physics. 2009. link Times cited: 39
Abstract: A reactive force field, REAXFF, for aluminum hydride has bee… read more
Abstract: A reactive force field, REAXFF, for aluminum hydride has been developed based on density functional theory (DFT) derived data. REAXFF(AlH(3)) is used to study the dynamics governing hydrogen desorption in AlH(3). During the abstraction process of surface molecular hydrogen charge transfer is found to be well described by REAXFF(AlH(3)). Results on heat of desorption versus cluster size show that there is a strong dependence of the heat of desorption on the particle size, which implies that nanostructuring enhances desorption process. In the gas phase, it was observed that small alane clusters agglomerated into a bigger cluster. After agglomeration molecular hydrogen was desorbed from the structure. This thermodynamically driven spontaneous agglomeration followed by desorption of molecular hydrogen provides a mechanism on how mobile alane clusters can facilitate the mass transport of aluminum atoms during the thermal decomposition of NaAlH(4). read less
NOT USED (high confidence) I. Schweigert and B. Dunlap, “Electronic structure and molecular dynamics of breaking the RO-NO2 bond.,” The Journal of chemical physics. 2009. link Times cited: 14
Abstract: Decomposition of energetic molecules such as pentaerythritol… read more
Abstract: Decomposition of energetic molecules such as pentaerythritol tetranitrate is accompanied by extensive changes in their electronic configuration and thus is challenging for ab initio Born-Oppenheimer molecular dynamics simulations. The performance of single-determinant methods (in particular, density-functional theory) is validated on electronic structure and molecular dynamics simulations of RO-NO(2) bond dissociation in a smaller nitric ester, ethyl nitrate. Accurate description of dissociating molecule requires using unrestricted, spin-symmetry-broken orbitals. However, the iterative self-consistent field procedure is prone to convergence failures in the bond-breaking region even if robust convergence algorithms are employed. As a result, molecular dynamics simulations of unimolecular decomposition need to be closely monitored and manually restarted to ensure seamless transition from the closed-shell to open-shell configuration. read less
NOT USED (high confidence) S. Zybin, W. G. III, P. Xu, J. Budzien, and A. Thompson, “Reactive Molecular Dynamics of Shock- and Shear-Induced Chemistry in Energetic Materials for Future Force Insensitive Munitions,” 2009 DoD High Performance Computing Modernization Program Users Group Conference. 2009. link Times cited: 2
Abstract: We report an approach to large-scale atomistic simulations o… read more
Abstract: We report an approach to large-scale atomistic simulations of chemical initiation processes in shocked energetic materials based on parallel implementation of the ReaxFF reactive force field. Here, we present results of reactive molecular dynamics (MD) simulations of shocked Pentaerythritol Tetranitrate (PETN) single crystal, a conventional high explosive. We study a planar wall impact to compare mechanical and chemical response at different speeds. The dominant initiation reactions in both systems lead to the formation of NO2. The lagging secondary reactions lead to a formation of water, nitrogen, and other products. By tracking the position of the shock front as a function of time, we have been able to observe how the shock velocity changes in response to the storage and release of chemical energy behind the shock front. We also investigate the effect of shear along different slip systems on chemical initiation. All calculations are performed with massively parallel MD code GRASP enabling multi-million atom reactive MD simulations of chemical processes in many important stockpile materials. read less
NOT USED (high confidence) T. Verstraelen et al., “Multi-level Modeling of Silica–Template Interactions During Initial Stages of Zeolite Synthesis,” Topics in Catalysis. 2009. link Times cited: 32
NOT USED (high confidence) C. J. Wu, L. Fried, L. Yang, N. Goldman, and S. Bastea, “Catalytic behaviour of dense hot water.,” Nature chemistry. 2009. link Times cited: 92
NOT USED (high confidence) M. Manaa, E. Reed, L. Fried, and N. Goldman, “Nitrogen-rich heterocycles as reactivity retardants in shocked insensitive explosives.,” Journal of the American Chemical Society. 2009. link Times cited: 179
Abstract: We report the first quantum-based multiscale simulations to … read more
Abstract: We report the first quantum-based multiscale simulations to study the reactivity of shocked perfect crystals of the insensitive energetic material triaminotrinitrobenzene (TATB). Tracking chemical transformations of TATB experiencing overdriven shock speeds of 9 km/s for up to 0.43 ns and 10 km/s for up to 0.2 ns reveal high concentrations of nitrogen-rich heterocyclic clusters. Further reactivity of TATB toward the final decomposition products of fluid N(2) and solid carbon is inhibited due to the formation of these heterocycles. Our results thus suggest a new mechanism for carbon-rich explosive materials that precedes the slow diffusion-limited process of forming the bulk solid from carbon clusters and provide fundamental insight at the atomistic level into the long reaction zone of shocked TATB. read less
NOT USED (high confidence) N. Goldman, E. Reed, I. W. Kuo, L. Fried, C. Mundy, and A. Curioni, “Ab initio simulation of the equation of state and kinetics of shocked water.,” The Journal of chemical physics. 2009. link Times cited: 91
Abstract: We report herein first principles simulations of water under… read more
Abstract: We report herein first principles simulations of water under shock loading and the chemical reactivity under these hot, compressed conditions. Using a recently developed simulation technique for shock compression, we observe that water achieves chemical equilibrium in less than 2 ps for all shock conditions studied. We make comparison to the experimental results for the Hugoniot pressure and density final states. Our simulations show that decomposition occurs through the reversible reaction H(2)O <--> H(+) + OH(-), in agreement with experiment. Near the approximate intersection of the Hugoniot and the Neptune isentrope, we observe high concentrations of charged species that contribute electronic states near the band gap. read less
NOT USED (high confidence) G. Kaupp, “Mechanochemistry: the varied applications of mechanical bond-breaking,” CrystEngComm. 2009. link Times cited: 322
Abstract: Mechanochemistry means mechanical breakage of intramolecular… read more
Abstract: Mechanochemistry means mechanical breakage of intramolecular bonds by external force and must be differentiated from molecular solid-state chemistry, where contacts between micronized molecular solids are created by the mechanical action for mutual approach of the reacting centers. After an outline of the mechanistic differences, the varied mechanochemistry is discussed. Grinding, milling, shearing, scratching, polishing, and rapid friction (for polymers also cutting, kneading, extruding) provide the mechanical impact for mechanochemistry, while sonication and shock waving for intramolecular bond breaking are generally described as thermal processes. The various types of mechanophysics (e.g., mechanoelectricity, conformational changes, thixotropy, rheopexy, stirring of Newtonian liquids or suspensions, etc.) are not treated here. Mechanochemistry covers solid-state reactions of infinitely covalent crystals, brittle metals, polymers, molecular solids with weak covalent bonds, strong intramolecular bond breakage in shearing Bridgman's anvil or by friction at lubrication of rapidly moving cold contacting surfaces, and single bond breaking or cutting. The diverse wealth of practical applications of mechanochemistry is outlined with typical examples for ceramics, mechanical alloying, hydrogen storage, organic syntheses, waste remediation, leachings, surface plasmas, radical formation, explosives, nanotube formation, nanoparticles grafting, polymer technology, radical initiation, scratch-less polishing, wear protection, lubrication, mechanochromism, nano-dissection, and many more. read less
NOT USED (high confidence) J. J. Ojwang, R. A. Santen, G. Kramer, A. V. Duin, and W. Goddard, “Predictions of melting, crystallization, and local atomic arrangements of aluminum clusters using a reactive force field.,” The Journal of chemical physics. 2008. link Times cited: 50
Abstract: A parametrized reactive force field model for aluminum ReaxF… read more
Abstract: A parametrized reactive force field model for aluminum ReaxFF(Al) has been developed based on density functional theory (DFT) data. A comparison has been made between DFT and ReaxFF(Al) outputs to ascertain whether ReaxFF(Al) is properly parametrized and to check if the output of the latter has correlation with DFT results. Further checks include comparing the equations of state of condensed phases of Al as calculated from DFT and ReaxFF(Al). There is a good match between the two results, again showing that ReaxFF(Al) is correctly parametrized as per the DFT input. Simulated annealing has been performed on aluminum clusters Al(n) using ReaxFF(Al) to find the stable isomers of the clusters. A plot of stability function versus cluster size shows the existence of highly stable clusters (magic clusters). Quantum mechanically these magic clusters arise due to the complete filling of the orbital shells. However, since force fields do not care about electrons but work on the assumption of validity of Born-Oppenheimer approximation, the magic clusters are therefore correlated with high structural symmetry. There is a rapid decline in surface energy contribution due to the triangulated nature of the surface atoms leading to higher coordination number. The bulk binding energy is computed to be 76.8 kcal/mol. This gives confidence in the suitability of ReaxFF for studying and understanding the underlying dynamics in aluminum clusters. In the quantification of the growth of cluster it is seen that as the size of the clusters increase there is preference for the coexistence of fcc/hcp orders at the expense of simple icosahedral ordering, although there is some contribution from distorted icosahedral ordering. It is found that even for aluminum clusters with 512 atoms distorted icosahedral ordering exists. For clusters with N>/=256 atoms fcc ordering dominates, which implies that at this point we are already on the threshold of bulklike bonding. read less
NOT USED (high confidence) E. Reed, M. Armstrong, K. Kim, and J. Glownia, “Atomic-scale time and space resolution of THz frequency acoustic waves,” LEOS 2008 - 21st Annual Meeting of the IEEE Lasers and Electro-Optics Society. 2008. link Times cited: 1
Abstract: Our simulations and experiments demonstrate a new physical m… read more
Abstract: Our simulations and experiments demonstrate a new physical mechanism for detecting acoustic waves of THz frequencies. Detectable THz-frequency radiation is generated when the acoustic wave passes a piezoelectric interface. read less
NOT USED (high confidence) M. Buehler, S. Keten, and T. Ackbarow, “Theoretical and computational hierarchical nanomechanics of protein materials: Deformation and fracture,” Progress in Materials Science. 2008. link Times cited: 179
NOT USED (high confidence) J. Xu, J. Zhao, and L. Sun, “Thermal decomposition behaviour of RDX by first-principles molecular dynamics simulation,” Molecular Simulation. 2008. link Times cited: 9
Abstract: Thermal decomposition behaviour of cyclotrimethylenetrinitra… read more
Abstract: Thermal decomposition behaviour of cyclotrimethylenetrinitramine crystal (with a density of 1.81 g/cm3 and at a temperature of 3000 K) was simulated using density functional molecular dynamics up to 33 ps. During the entire simulation time, the major products are N2, H2O and CO2; and their populations generally increase with time. In the initial stage of decomposition, we observed formation of NO2 groups carrying about one positive charge, which might play some roles in the further decomposition process. The energy transformation during the thermal decomposition process is also discussed. read less
NOT USED (high confidence) W. Goddard, K. Chenoweth, S. Pudar, A. Duin, and M. Cheng, “Structures, Mechanisms, and Kinetics of Selective Ammoxidation and Oxidation of Propane over Multi-metal Oxide Catalysts,” Topics in Catalysis. 2008. link Times cited: 52
NOT USED (high confidence) A. A. Selezenev, A. Aleinikov, and I. Briginas, “A molecular dynamics simulation of the destruction of explosive molecules at high-velocity collisions,” Russian Journal of Physical Chemistry B. 2008. link Times cited: 2
NOT USED (high confidence) J. J. Ojwang, R. A. Santen, G. Kramer, A. V. Duin, and W. Goddard, “Modeling the sorption dynamics of NaH using a reactive force field.,” The Journal of chemical physics. 2008. link Times cited: 32
Abstract: We have parametrized a reactive force field for NaH, ReaxFF(… read more
Abstract: We have parametrized a reactive force field for NaH, ReaxFF(NaH), against a training set of ab initio derived data. To ascertain that ReaxFF(NaH) is properly parametrized, a comparison between ab initio heats of formation of small representative NaH clusters with ReaxFF(NaH) was done. The results and trend of ReaxFF(NaH) are found to be consistent with ab initio values. Further validation includes comparing the equations of state of condensed phases of Na and NaH as calculated from ab initio and ReaxFF(NaH). There is a good match between the two results, showing that ReaxFF(NaH) is correctly parametrized by the ab initio training set. ReaxFF(NaH) has been used to study the dynamics of hydrogen desorption in NaH particles. We find that ReaxFF(NaH) properly describes the surface molecular hydrogen charge transfer during the abstraction process. Results on heat of desorption versus cluster size shows that there is a strong dependence on the heat of desorption on the particle size, which implies that nanostructuring enhances desorption process. To gain more insight into the structural transformations of NaH during thermal decomposition, we performed a heating run in a molecular dynamics simulation. These runs exhibit a series of drops in potential energy, associated with cluster fragmentation and desorption of molecular hydrogen. This is consistent with experimental evidence that NaH dissociates at its melting point into smaller fragments. read less
NOT USED (high confidence) F. Shimojo, R. Kalia, A. Nakano, and P. Vashishta, “Divide-and-conquer density functional theory on hierarchical real-space grids: Parallel implementation and applications,” Physical Review B. 2008. link Times cited: 60
Abstract: A linear-scaling algorithm based on a divide-and-conquer ! D… read more
Abstract: A linear-scaling algorithm based on a divide-and-conquer ! DC" scheme has been designed to perform large-scale molecular-dynamics ! MD" simulations, in which interatomic forces are computed quantum mechanically in the framework of the density functional theory ! DFT" . Electronic wave functions are represented on a real-space grid, which is augmented with a coarse multigrid to accelerate the convergence of iterative solutions and with adaptive fine grids around atoms to accurately calculate ionic pseudopotentials. Spatial decomposition is employed to implement the hierarchical-grid DC-DFT algorithm on massively parallel computers. The largest benchmark tests include 11.8! 10 6 -atom ! 1.04! 10 12 electronic degrees of freedom" calculation on 131 072 IBM BlueGene/L processors. The DC-DFT algorithm has well-defined parameters to control the data locality, with which the solutions converge rapidly. Also, the total energy is well conserved during the MD simulation. We perform first-principles MD simulations based on the DC-DFT algorithm, in which large system sizes bring in excellent agreement with x-ray scattering measurements for the pairdistribution function of liquid Rb and allow the description of low-frequency vibrational modes of graphene. The band gap of a CdSe nanorod calculated by the DC-DFT algorithm agrees well with the available conventional DFT results. With the DC-DFT algorithm, the band gap is calculated for larger system sizes until the result reaches the asymptotic value. DOI: 10.1103/PhysRevB.77.085103 read less
NOT USED (high confidence) K. Nomura, R. Kalia, A. Nakano, and P. Vashishta, “A scalable parallel algorithm for large-scale reactive force-field molecular dynamics simulations,” Comput. Phys. Commun. 2008. link Times cited: 77
NOT USED (high confidence) D. Mathieu, “Split charge equilibration method with correct dissociation limits.,” The Journal of chemical physics. 2007. link Times cited: 39
Abstract: Analytic reactive potentials rely on electronegativity equal… read more
Abstract: Analytic reactive potentials rely on electronegativity equalization to describe how the electron distribution is affected as chemical reactions occur. However, such models predict fractional charges for neutral species with different electronegativities. To overcome this well-known dissociation problem, an approach taking advantage of the concept of split charges [R. A. Nistor, J. G. Polihronov, M. H. Muser, and N. J. Mosey, J. Chem. Phys. 125, 094108 (2006)] is put forward. A first implementation is presented. Starting from a previous model [P. Bultinck, W. Langenaeker, P. Lahorte, F. D. Proft, P. Geerlings, M. Waroquier, and J. P. Tollenaere, J. Phys. Chem. A 106, 7887 (2002)], a new contribution to the total energy is introduced in order to make up for the lack of suitable constraints on the charge density. Its effect is to restrain charge transfer between remote atoms. As a consequence, systems in gas phase naturally decompose into neutral fragments. This result is achieved using two empirical parameters in addition to atomic electronegativities and hardnesses. read less
NOT USED (high confidence) K. Nomura, R. Kalia, A. Nakano, P. Vashishta, A. V. van Duin, and W. Goddard, “Dynamic transition in the structure of an energetic crystal during chemical reactions at shock front prior to detonation.,” Physical review letters. 2007. link Times cited: 116
Abstract: Mechanical stimuli in energetic materials initiate chemical … read more
Abstract: Mechanical stimuli in energetic materials initiate chemical reactions at shock fronts prior to detonation. Shock sensitivity measurements provide widely varying results, and quantum-mechanical calculations are unable to handle systems large enough to describe shock structure. Recent developments in reactive force-field molecular dynamics (ReaxFF-MD) combined with advances in parallel computing have paved the way to accurately simulate reaction pathways along with the structure of shock fronts. Our multimillion-atom ReaxFF-MD simulations of l,3,5-trinitro-l,3,5-triazine (RDX) reveal that detonation is preceded by a transition from a diffuse shock front with well-ordered molecular dipoles behind it to a disordered dipole distribution behind a sharp front. read less
NOT USED (high confidence) Y. Shi and D. Brenner, “Simulated thermal decomposition and detonation of nitrogen cubane by molecular dynamics.,” The Journal of chemical physics. 2007. link Times cited: 18
Abstract: We present simulations of a model molecular solid of nitroge… read more
Abstract: We present simulations of a model molecular solid of nitrogen cubane subject to thermal agitation and mechanical shock. A new approach, a reactive state summation potential, has been used to model nitrogen cubane dissociation. At elevated temperatures, the system decomposes to N(2) mixed with a small amount of oligomeric nitrogen. When subject to shock loading the system detonates above some critical threshold after which a shock front is self-sustained by the energy release from chemical reactions at a constant intrinsic speed. This is the first example of a fully three-dimensional atomic simulation of a chemically-sustained detonation. The spatial confinement of the shock front results in longer chain intermediates than in the case of thermal decomposition, suggesting that shock intermediates can be structurally very different from the same material subject to comparable temperatures and pressures. read less
NOT USED (high confidence) N. Umezawa, R. Kalia, A. Nakano, P. Vashista, and F. Shimojo, “1,3,5-trinitro-1,3,5-triazine decomposition and chemisorption on Al(111) surface: first-principles molecular dynamics study.,” The Journal of chemical physics. 2007. link Times cited: 31
Abstract: We have investigated the decomposition and chemisorption of … read more
Abstract: We have investigated the decomposition and chemisorption of a 1,3,5-trinitro-1,3,5-triazine (RDX) molecule on Al(111) surface using molecular dynamics simulations, in which interatomic forces are computed quantum mechanically in the framework of the density functional theory (DFT). The real-space DFT calculations are based on higher-order finite difference and norm-conserving pseudopotential methods. Strong attractive forces between oxygen and aluminum atoms break N-O and N-N bonds in the RDX and, subsequently, the dissociated oxygen atoms and NO molecules oxidize the Al surface. In addition to these Al surface-assisted decompositions, ring cleavage of the RDX molecule is also observed. These reactions occur spontaneously without potential barriers and result in the attachment of the rest of the RDX molecule to the surface. This opens up the possibility of coating Al nanoparticles with RDX molecules to avoid the detrimental effect of oxidation in high energy density material applications. read less
NOT USED (high confidence) A. Nakano et al., “A Divide-and-Conquer/Cellular-Decomposition Framework for Million-to-Billion Atom Simulations of Chemical Reactions,” Computational Materials Science. 2007. link Times cited: 92
NOT USED (high confidence) J. Fish, “Bridging the scales in nano engineering and science,” Journal of Nanoparticle Research. 2006. link Times cited: 156
NOT USED (high confidence) M. Buehler, “Atomistic and continuum modeling of mechanical properties of collagen: Elasticity, fracture, and self-assembly,” Journal of Materials Research. 2006. link Times cited: 268
Abstract: We report studies of the mechanical properties of tropocolla… read more
Abstract: We report studies of the mechanical properties of tropocollagen molecules under different types of mechanical loading including tension, compression, shear, and bending. Our modeling yields predictions of the fracture strength of single tropocollagen molecules and polypeptides, and also allows for quantification of the interactions between tropocollagen molecules. Atomistic modeling predicts a persistence length of tropocollagen molecules ξ ≈ 23.4 nm, close to experimental measurements. Our studies suggest that to describe large-strain or hyperelastic properties, it is critical to include a correct description of the bond behavior and breaking processes at large bond stretch, information that stems from the quantum chemical details of bonding. We use full atomistic calculations to derive parameters for a mesoscopic bead-spring model of tropocollagen molecules. We demonstrate that the mesoscopic model enables one to study the finite temperature, long-time scale behavior of tropocollagen fibers, illustrating the dynamics of solvated tropocollagen molecules for different molecular lengths. read less
NOT USED (high confidence) W. Goddard et al., “Development of the ReaxFF reactive force field for mechanistic studies of catalytic selective oxidation processes on BiMoOx,” Topics in Catalysis. 2006. link Times cited: 94
NOT USED (high confidence) S. Boyd, M. Gravelle, and P. Politzer, “Nonreactive molecular dynamics force field for crystalline hexahydro-1,3,5-trinitro-1,3,5 triazine.,” The Journal of chemical physics. 2006. link Times cited: 37
Abstract: An empirical nonreactive force field has been developed for … read more
Abstract: An empirical nonreactive force field has been developed for molecular dynamics (MD)/Monte Carlo simulation of the formation, diffusion, and agglomeration of point defects in the crystal lattice of the alpha modification of hexahydro-1,3,5-trinitro-1,3,5 triazine (RDX) using flexible molecules. Bond stretching and angle bending are represented by Morse and harmonic functions, and torsion by a truncated cosine series. Nonbonded interactions, both inter- and intramolecular, are described by Buckingham potentials separately parametrized. Intermolecular electrostatic interactions are treated via a Coulomb term coupled with a smooth 15.0 A cutoff radius. Parameters were taken in part from earlier published works and were determined partly by fitting to known molecular and crystal properties of RDX. In MD simulations at constant pressure and temperature, the model was able to stabilize and maintain the correct crystal structure, symmetry, and molecular conformation of alpha-RDX. Vibrational frequencies, lattice binding energy and dimensions, coefficients of thermal expansion, and several unusually short intermolecular distances are all reproduced in satisfactory agreement with experimental data. read less
NOT USED (high confidence) M. Buehler, A. V. van Duin, and W. Goddard, “Multiparadigm modeling of dynamical crack propagation in silicon using a reactive force field.,” Physical review letters. 2006. link Times cited: 199
Abstract: We report a study of dynamic cracking in a silicon single cr… read more
Abstract: We report a study of dynamic cracking in a silicon single crystal in which the ReaxFF reactive force field is used for several thousand atoms near the crack tip, while more than 100,000 atoms are described with a nonreactive force field. ReaxFF is completely derived from quantum mechanical calculations of simple silicon systems without any empirical parameters. Our results reproduce experimental observations of fracture in silicon including changes in crack dynamics for different crack orientations. read less
NOT USED (high confidence) W. Goddard et al., “Multi-paradigm multi-scale simulations for fuel cell catalysts and membranes,” Molecular Simulation. 2006. link Times cited: 115
Abstract: Dramatically improving the performance of fuel cell systems … read more
Abstract: Dramatically improving the performance of fuel cell systems with their complex heterogeneous structures involving electrocatalysts, proton conducting membrane, reactant, and interfaces between them requires understanding the fundamental chemical, electrochemical, and physical phenomena at the heart of these complex materials and relating these fundamentals to the properties and performance of the membrane–electrode assembly. Our goal is to develop a predictive model that can be used to estimate the changes in performance upon changes in the design and which can be used to monitor performance of working fuel cells. Our strategy is to start with first principles quantum mechanics (QM) and to develop overlapping simulation methodologies in which QM is used to train a reactive force field that can be applied for large-scale (millions of atom) molecular dynamics simulations while retaining the accuracy of QM. The results of molecular dynamics are used to extract a coarse grain or mesoscale description useful in modeling properties at much larger scales. This model would enable the conception, synthesis, fabrication, characterization, and development of advanced materials and structures for fuel cells and for the associated hydrocarbon fuel reformers in an overall fuel cell system. We illustrate here some of the progress toward this goal. read less
NOT USED (high confidence) S. Han, J. Kang, H.-M. Lee, A. Duin, and W. Goddard, “The theoretical study on interaction of hydrogen with single-walled boron nitride nanotubes. I. The reactive force field ReaxFFHBN development,” Journal of Chemical Physics. 2005. link Times cited: 58
Abstract: We present a new reactive force field ReaxFFHBN derived to a… read more
Abstract: We present a new reactive force field ReaxFFHBN derived to accurately model large molecular and condensed phase systems of H, B, and N atoms. ReaxFFHBN has been tested against quantum calculation data for B–H, B–B, and B–N bond dissociations and for H–B–H, B–N–B, and N–B–N bond angle strain energies of various molecular clusters. The accuracy of the developed ReaxFFHBN for B–N–H systems is also tested for (i) H–B and H–B bond energies as a function of out of plane in H–B(NH2)3 and H–N(BH2)3, respectively, (ii) the reaction energy for the B3N3H6+H2-->B3N3H8, and (iii) crystal properties such as lattice parameters and equations of states for the hexagonal type (h-BN) with a graphite structure and for the cubic type (c-BN) with a zinc-blende structure. For all these systems, ReaxFFHBN gives reliable results consistent with those from quantum calculations as it describes well bond breaking and formation in chemical processes and physical properties. Consequently, the molecular-dynamics simulation based on ReaxFFHBN is expected to give a good description of large systems (>2000 atoms even on the one-CPU machine) with hydrogen, boron, and nitrogen atoms. read less
NOT USED (high confidence) S. Han, J. Kang, H.-M. Lee, A. Duin, and W. Goddard, “Liquefaction of H2 molecules upon exterior surfaces of carbon nanotube bundles,” Applied Physics Letters. 2005. link Times cited: 30
Abstract: We have used molecular dynamics simulations to investigate i… read more
Abstract: We have used molecular dynamics simulations to investigate interaction of H2 molecules on the exterior surfaces of carbon nanotubes (CNTs): single and bundle types. At 80 K and 10 MPa, it is found that charge transfer occurs from a low curvature region to a high curvature region of the deformed CNT bundle, which develops charge polarization only on the deformed structure. The long-range electrostatic interactions of polarized charges on the deformed CNT bundle with hydrogen molecules are observed to induce a high local-ordering of H2 gas that results in hydrogen liquefaction. Our predicted heat of hydrogen liquefaction on the CNT bundle is 97.6 kcal kg^-1. On the other hand, hydrogen liquefaction is not observed in the CNT of a single type. This is because charge polarization is not developed on the single CNT as it is symmetrically deformed under the same pressure. Consequently, the hydrogen storage capacity on the CNT bundle is much higher due to liquefaction than that on the single CNT. Additionally, our results indicate that it would also be possible to liquefy H2 gas on a more strongly polarized CNT bundle at temperatures higher than 80 K. read less
NOT USED (high confidence) F. Shimojo, R. Kalia, A. Nakano, and P. Vashishta, “Embedded divide-and-conquer algorithm on hierarchical real-space grids: parallel molecular dynamics simulation based on linear-scaling density functional theory,” Comput. Phys. Commun. 2005. link Times cited: 65
NOT USED (high confidence) A. Strachan, E. Kober, A. V. van Duin, J. Oxgaard, and W. Goddard, “Thermal decomposition of RDX from reactive molecular dynamics.,” The Journal of chemical physics. 2005. link Times cited: 356
Abstract: We use the recently developed reactive force field ReaxFF wi… read more
Abstract: We use the recently developed reactive force field ReaxFF with molecular dynamics to study thermal induced chemistry in RDX [cyclic-[CH(2)N(NO(2))](3)] at various temperatures and densities. We find that the time evolution of the potential energy can be described reasonably well with a single exponential function from which we obtain an overall characteristic time of decomposition that increases with decreasing density and shows an Arrhenius temperature dependence. These characteristic timescales are in reasonable quantitative agreement with experimental measurements in a similar energetic material, HMX [cyclic-[CH(2)N(NO(2))](4)]. Our simulations show that the equilibrium population of CO and CO(2) (as well as their time evolution) depend strongly of density: at low density almost all carbon atoms form CO molecules; as the density increases larger aggregates of carbon appear leading to a C deficient gas phase and the appearance of CO(2) molecules. The equilibrium populations of N(2) and H(2)O are more insensitive with respect to density and form in the early stages of the decomposition process with similar timescales. read less
NOT USED (high confidence) A. Lipanov, N. V. Khokhryakov, and V. Kodolov, “Mathematical Modeling of Octogen Destruction,” Doklady Physical Chemistry. 2004. link Times cited: 0
NOT USED (high confidence) M. Manaa, L. Fried, and E. Reed, “Explosive chemistry: Simulating the chemistry of energetic materials at extreme conditions,” Journal of Computer-Aided Materials Design. 2003. link Times cited: 30
NOT USED (high confidence) K. Lynch, A. Thompson, and A. Strachan, “Coarse grain modeling of spall failure in molecular crystals: role of intra-molecular degrees of freedom,” Modelling and Simulation in Materials Science and Engineering. 2008. link Times cited: 21
Abstract: We use a recently developed thermodynamically accurate mesod… read more
Abstract: We use a recently developed thermodynamically accurate mesodynamical method (Strachan and Holian 2005 Phys. Rev. Lett. 94 014301) where groups of atoms are represented by mesoparticles to characterize the shock compression and dynamical failure (spall) of a model molecular crystal. We characterize how the temperature rise caused by the shockwave depends on the specific heat of the degrees of freedom (DoFs) internal to the mesoparticles (Cint) and the strength of the coupling between the internal DoFs and the mesoparticles. We find that the temperature of the shocked material decreases with increasing Cint and decreasing coupling and quantify these effects. Our simulations also show that the threshold for plastic deformation (the Hugoniot elastic limit) depends on the properties of the internal DoFs while the threshold for failure is very insensitive to them. These results have implications on the results of all-atom MD simulations, whose classical nature leads to a significant overestimation of the specific heat of molecular materials. read less
NOT USED (high confidence) B. Rice, E. Byrd, and W. Mattson, “Computational Aspects of Nitrogen-Rich HEDMs.” 2007. link Times cited: 52
NOT USED (high confidence) M. Buehler, J. Dodson, A. Duin, and W. Goddard, “The Computational Materials Design Facility (CMDF): A powerful framework for multi-paradigm multi-scale simulations,” MRS Proceedings. 2005. link Times cited: 22
Abstract: Predicting the properties and behavior of materials by compu… read more
Abstract: Predicting the properties and behavior of materials by computer simulation from a fundamental, ab initio perspective has long been a vision of computational material scientists. The key to achieving this goal is utilizing hierarchies of paradigms and scales that connect macrosystems to first principles quantum mechanics (QM). Here we describe a new software environment, the “Computational Materials Design Facility” (CMDF), capable of simulations of complex materials studies using a variety of simulation paradigms. The CMDF utilizes a Python scripting layer to integrate different computational tools to develop multi-scale simulation applications. We have integrated DFT QM methods, the first principles ReaxFF reactive force field, empirical all atom force fields (FFs), mesoscale and continuum methods. The central data structure Extended OpenBabel (XOB) plays a critical role as glue between applications. We demonstrate the usefulness of CMDF in examples that couple complex chemistry and mechanical properties during dynamical failure processes, as for example in a study of cracking of Ni under presence of O 2 . read less
NOT USED (definite) Y. Zhang, H. Liu, Z. Yang, Q. Li, and Y. He, “Shock-Induced Hot Spot Formation and Spalling in 1,3,5-trinitroperhydro-1,3,5-triazine Containing a Cube Void,” ACS Omega. 2019. link Times cited: 6
Abstract: The initial reaction mechanism of energetic materials under … read more
Abstract: The initial reaction mechanism of energetic materials under impact loading and the role of crystal properties in impact initiation and sensitivity are still unclear. In this paper, we report reactive molecular dynamics simulations of shock initiation of 1,3,5-trinitroperhydro-1,3,5-triazine (RDX) crystals containing a cube void. Shock-induced void collapse, hot spots formation and growth, as well as spalling are revealed to be dependent on the shock velocity. The void collapse times are 1.5 and 0.7 ps, for the shock velocity of 2 and 4 km·s–1, respectively. Results indicate that the initial hot spot formation consists of two steps: one is the temperature rise caused by local plastic deformation and the other is the temperature increase resulting from the collision of upstream and downstream particles during the void collapse. Whether hot spots will continue to grow or quench depends on sensitive balance between energy release caused by local physical and chemical reactions and various heat dissipation mechanisms. In our simulations, hot spot would grow for Up = 4 km·s–1; hot spot is weak to some extent for Up = 2 km·s–1. The tensile wave reflected by the shock wave after reaching the free surface causes the spalling, which depends on the initial shock velocity. Typical spalling occurs for the shock velocity 2 km·s–1, while the tensile wave induces the microsplit region in RDX crystals in the case of Up = 4 km·s–1. Chemical reactions are studied for Rankine–Hugoniot shock pressures Ps = 14.4, 57.8 GPa. For the weak shock, there is almost no decomposition reaction of the RDX molecules near the spalling region. On the contrary, there are large number of small molecule products, such as H2O, CO2, NO2, and so forth, around the microsplit regions for the strong shock. The ruptures of N–NO2 bond are the main initial reaction mechanisms for the shocked RDX crystal and are not affected by shock strength, while the microsplit slows down the decomposition rate of RDX. The work in this paper can shed light on a thorough understanding of thermal ignition, hot spot growth, and other physical and chemical phenomena of energetic materials containing voids under impact loading. read less
NOT USED (definite) A. Mishra et al., “Multiobjective genetic training and uncertainty quantification of reactive force fields,” npj Computational Materials. 2018. link Times cited: 25
NOT USED (definite) M. Radue, B. D. Jensen, S. Gowtham, D. R. Klimek-McDonald, J. King, and G. Odegard, “Comparing the Mechanical Response of Di-, Tri-, and Tetra-functional Resin Epoxies with Reactive Molecular Dynamics.,” Journal of polymer science. Part B, Polymer physics. 2018. link Times cited: 50
Abstract: The influence of monomer functionality on the mechanical pro… read more
Abstract: The influence of monomer functionality on the mechanical properties of epoxies is studied using Molecular Dynamics (MD) with the Reax Force Field (ReaxFF). From deformation simulations, the Young's modulus, yield point, and Poisson's ratio are calculated and analyzed. The results demonstrate an increase in stiffness and yield strength with increasing resin functionality. Comparison between the network structures of distinct epoxies is further advanced by the Monomeric Degree Index (MDI). Experimental validation demonstrates the MD results correctly predict the relationship in Young's moduli. Therefore, ReaxFF is confirmed to be a useful tool for studying the mechanical behavior of epoxies. read less
NOT USED (definite) H. Liu, P. Zhou, H. Li, A. Li, and Y. Dou, “DFTB-MD Simulation of Shocked Water Cluster.” 2016. link Times cited: 0
Abstract: Relatively efficient and precise quantum molecular dynamics … read more
Abstract: Relatively efficient and precise quantum molecular dynamics simulations were performed to gain fundamental insights into the mechanisms for the primary detonation process of water under shock wave loading using self-consistent charge density-functional tight binding (SCC-DFTB) calculations combined with the multiscale shock technique (MSST) as well as DFTB+ program conjunct with MSST. We observe that water achieves chemical equilibrium in less than 4ps for all shock conditions studied. What's more, we make comparison with the experimental results for the Hugoniot pressure and density final states. At last, our simulations show that decomposition occurs through the reversible H2O↔H + +OH - , in agreement with experiment. Therefore, the molecular dynamics method of water under extreme conditions is effective. read less
NOT USED (definite) T. Senftle et al., “The ReaxFF reactive force-field: development, applications and future directions.” 2016. link Times cited: 1212
NOT USED (definite) Q. An, T. Cheng, W. Goddard, and S. Zybin, “Anisotropic Impact Sensitivity and Shock Induced Plasticity of TKX-50 (Dihydroxylammonium 5,5′-bis(tetrazole)-,1′-diolate) Single Crystals: From Large-Scale Molecular Dynamics Simulations,” Journal of Physical Chemistry C. 2015. link Times cited: 58
Abstract: Dihydroxylammonium 5,5′-bis(tetrazole)-1,1′-diolate (TKX-50)… read more
Abstract: Dihydroxylammonium 5,5′-bis(tetrazole)-1,1′-diolate (TKX-50) is a newly synthesized energetic material with high energy storage, low impact sensitivity, and low toxicity. These features make it a viable candidate to replace such commonly used energetic materials as RDX and CL-20 in the next generation of explosives. Sensitivity determines the engineering application of energetic materials (EMs) and has been widely studied for various EMs. To understand the origin of the anisotropic sensitivity and properties of this new synthesized EM, we report a flexible classical force field for TKX-50 developed to reproduce the molecular properties (geometry, vibrational frequencies and torsion barriers) and the crystal properties (cell parameters and lattice energy). We then used this force field in molecular dynamics (MD) simulations to predict such thermodynamic and mechanical properties as isothermal compressibility, thermal expansion, elastic moduli, and heat capacity. Furthermore, we carried out large scale (∼a half million atoms) MD simulations to investigate the mechanical response to shocks in the [100], [010] and [001] directions. The predicted Hugoniot elastic limits (HELs) are 6.1 GPa for [100], 14.2 GPa for [010] and 9.1 GPa for [001] shocks. Thus, single crystal TKX-50 shows anisotropic impact sensitivity with [010] as the most sensitive direction and [100] as least sensitive. The plastic deformations in shock compression along the [100] direction primary arise from the (001)/[210] and (010)/[001] slip systems of. For the [010] shock, the primary slip systems are (100)/[021] and (001)/[210]. However, no obvious slip system was observed for [001] shock. read less
NOT USED (definite) M. Am-Shallem, Y. Zeiri, S. Zybin, and R. Kosloff, “Molecular dynamics simulations of weak detonations.,” Physical review. E, Statistical, nonlinear, and soft matter physics. 2011. link Times cited: 3
Abstract: Detonation of a three-dimensional reactive nonisotropic mole… read more
Abstract: Detonation of a three-dimensional reactive nonisotropic molecular crystal is modeled using molecular dynamics simulations. The detonation process is initiated by an impulse, followed by the creation of a stable fast reactive shock wave. The terminal shock velocity is independent of the initiation conditions. Further analysis shows supersonic propagation decoupled from the dynamics of the decomposed material left behind the shock front. The dependence of the shock velocity on crystal nonlinear compressibility resembles solitary behavior. These properties categorize the phenomena as a weak detonation. The dependence of the detonation wave on microscopic potential parameters was investigated. An increase in detonation velocity with the reaction exothermicity reaching a saturation value is observed. In all other respects the model crystal exhibits typical properties of a molecular crystal. read less
NOT USED (definite) M. Buehler and S. Keten, “Colloquium: Failure of molecules, bones, and the Earth itself,” Reviews of Modern Physics. 2010. link Times cited: 40
Abstract: Laboratory for Atomistic and Molecular Mechanics, Department… read more
Abstract: Laboratory for Atomistic and Molecular Mechanics, Department of Civil and Environmental Engineering, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Room 1-235AB Center for Materials Science and Engineering, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, Massachusetts 02139, USA; and Center for Computational Engineering, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, Massachusetts 02139, USA read less
NOT USED (definite) A. Nakano et al., “De Novo Ultrascale Atomistic Simulations On High-End Parallel Supercomputers,” The International Journal of High Performance Computing Applications. 2008. link Times cited: 50
Abstract: We present a de novo hierarchical simulation framework for f… read more
Abstract: We present a de novo hierarchical simulation framework for first-principles based predictive simulations of materials and their validation on high-end parallel supercomputers and geographically distributed clusters. In this framework, high-end chemically reactive and non-reactive molecular dynamics (MD) simulations explore a wide solution space to discover microscopic mechanisms that govern macroscopic material properties, into which highly accurate quantum mechanical (QM) simulations are embedded to validate the discovered mechanisms and quantify the uncertainty of the solution. The framework includes an embedded divide-and-conquer (EDC) algorithmic framework for the design of linear-scaling simulation algorithms with minimal bandwidth complexity and tight error control. The EDC framework also enables adaptive hierarchical simulation with automated model transitioning assisted by graph-based event tracking. A tunable hierarchical cellular decomposition parallelization framework then maps the O(N) EDC algorithms onto petaflops computers, while achieving performance tunability through a hierarchy of parameterized cell data/ computation structures, as well as its implementation using hybrid grid remote procedure call + message passing + threads programming. High-end computing platforms such as IBM BlueGene/L, SGI Altix 3000 and the NSF TeraGrid provide an excellent test grounds for the framework. On these platforms, we have achieved unprecedented scales of quantum-mechanically accurate and well validated, chemically reactive atomistic simulations—1.06 billion-atom fast reactive force-field MD and 11.8 million-atom (1.04 trillion grid points) quantum-mechanical MD in the framework of the EDC density functional theory on adaptive multigrids— in addition to 134 billion-atom non-reactive space—time multiresolution MD, with the parallel efficiency as high as 0.998 on 65,536 dual-processor BlueGene/L nodes. We have also achieved an automated execution of hierarchical QM/MD simulation on a grid consisting of 6 supercomputer centers in the US and Japan (in total of 150,000 processor hours), in which the number of processors change dynamically on demand and resources are allocated and migrated dynamically in response to faults. Furthermore, performance portability has been demonstrated on a wide range of platforms such as BlueGene/L, Altix 3000, and AMD Opteron-based Linux clusters. read less
The long form of the KIM ID including a human readable prefix (100 characters max), two underscores, and the Short KIM ID. Extended KIM IDs can only contain alpha-numeric characters (letters and digits) and underscores and must begin with a letter.
Periodic boundary conditions were NOT supported correctly for at least one configuration that the model was able to compute. This is an error in the implementation of the model.
vc-periodicity-support
mandatory
Periodic boundary conditions are handled correctly; see full description.
Model energy does NOT have permutation symmetry for at least one configuration that the model was able to compute. This indicates an error in the implementation of the model.
vc-permutation-symmetry
mandatory
Total energy and forces are unchanged when swapping atoms of the same species; see full description.
The letter grade D was assigned because the normalized error in the computation was 2.11445e-02 compared with a machine precision of 2.22045e-16. The letter grade was based on 'score=log10(error/eps)', with ranges A=[0, 7.5], B=(7.5, 10.0], C=(10.0, 12.5], D=(12.5, 15.0), F>15.0. 'A' is the best grade, and 'F' indicates failure.
vc-forces-numerical-derivative
consistency
Forces computed by the model agree with numerical derivatives of the energy; see full description.
The model is C^0 continuous. This means that the model has continuous energy, but a discontinuous first derivative.
vc-dimer-continuity-c1
informational
The energy versus separation relation of a pair of atoms is C1 continuous (i.e. the function and its first derivative are continuous); see full description.
Model energy and/or forces are NOT invariant with respect to rigid-body translation and/or rotation for at least one configuration that the model was able to compute. This could be valid if the model includes an external field. Otherwise this is an error in the model implementation.
vc-objectivity
informational
Total energy is unchanged and forces transform correctly under rigid-body translation and rotation; see full description.
Model energy does NOT have inversion symmetry for at least one configuration that the model was able to compute. This could be valid if the model includes an external field or represents a material with unusual quantum properties. Otherwise this is an error in the model implementation.
vc-inversion-symmetry
informational
Total energy is unchanged and forces change sign when inverting a configuration through the origin; see full description.
The model returns the same energy and forces when computed in serial and when using parallel threads for a set of configurations. Note that this is not a guarantee of thread safety; see full description.
This bar chart plot shows the mono-atomic body-centered cubic (bcc) lattice constant predicted by the current model (shown in the unique color) compared with the predictions for all other models in the OpenKIM Repository that support the species. The vertical bars show the average and standard deviation (one sigma) bounds for all model predictions. Graphs are generated for each species supported by the model.
This graph shows the cohesive energy versus volume-per-atom for the current mode for four mono-atomic cubic phases (body-centered cubic (bcc), face-centered cubic (fcc), simple cubic (sc), and diamond). The curve with the lowest minimum is the ground state of the crystal if stable. (The crystal structure is enforced in these calculations, so the phase may not be stable.) Graphs are generated for each species supported by the model.
This bar chart plot shows the mono-atomic face-centered diamond lattice constant predicted by the current model (shown in the unique color) compared with the predictions for all other models in the OpenKIM Repository that support the species. The vertical bars show the average and standard deviation (one sigma) bounds for all model predictions. Graphs are generated for each species supported by the model.
This graph shows the dislocation core energy of a cubic crystal at zero temperature and pressure for a specific set of dislocation core cutoff radii. After obtaining the total energy of the system from conjugate gradient minimizations, non-singular, isotropic and anisotropic elasticity are applied to obtain the dislocation core energy for each of these supercells with different dipole distances. Graphs are generated for each species supported by the model.
This bar chart plot shows the mono-atomic face-centered cubic (fcc) elastic constants predicted by the current model (shown in blue) compared with the predictions for all other models in the OpenKIM Repository that support the species. The vertical bars show the average and standard deviation (one sigma) bounds for all model predictions. Graphs are generated for each species supported by the model.
This bar chart plot shows the mono-atomic face-centered cubic (fcc) lattice constant predicted by the current model (shown in red) compared with the predictions for all other models in the OpenKIM Repository that support the species. The vertical bars show the average and standard deviation (one sigma) bounds for all model predictions. Graphs are generated for each species supported by the model.
This bar chart plot shows the intrinsic and extrinsic stacking fault energies as well as the unstable stacking and unstable twinning energies for face-centered cubic (fcc) predicted by the current model (shown in blue) compared with the predictions for all other models in the OpenKIM Repository that support the species. The vertical bars show the average and standard deviation (one sigma) bounds for all model predictions. Graphs are generated for each species supported by the model.
This bar chart plot shows the mono-atomic face-centered cubic (fcc) relaxed surface energies predicted by the current model (shown in blue) compared with the predictions for all other models in the OpenKIM Repository that support the species. The vertical bars show the average and standard deviation (one sigma) bounds for all model predictions. Graphs are generated for each species supported by the model.
This bar chart plot shows the mono-atomic simple cubic (sc) lattice constant predicted by the current model (shown in the unique color) compared with the predictions for all other models in the OpenKIM Repository that support the species. The vertical bars show the average and standard deviation (one sigma) bounds for all model predictions. Graphs are generated for each species supported by the model.
This Test Driver uses LAMMPS to compute the cohesive energy of a given monoatomic cubic lattice (fcc, bcc, sc, or diamond) at a variety of lattice spacings. The lattice spacings range from a_min (=a_min_frac*a_0) to a_max (=a_max_frac*a_0) where a_0, a_min_frac, and a_max_frac are read from stdin (a_0 is typically approximately equal to the equilibrium lattice constant). The precise scaling and number of lattice spacings sampled between a_min and a_0 (a_0 and a_max) is specified by two additional parameters passed from stdin: N_lower and samplespacing_lower (N_upper and samplespacing_upper). Please see README.txt for further details.
Test
Test Results
Link to Test Results page
Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI)
Computes the elastic constants for an arbitrary crystal. A robust computational protocol is used, attempting multiple methods and step sizes to achieve an acceptably low error in numerical differentiation and deviation from material symmetry. The crystal structure is specified using the AFLOW prototype designation as part of the Crystal Genome testing framework. In addition, the distance from the obtained elasticity tensor to the nearest isotropic tensor is computed.
Test
Test Results
Link to Test Results page
Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI)
Computes the cubic elastic constants for some common crystal types (fcc, bcc, sc, diamond) by calculating the hessian of the energy density with respect to strain. An estimate of the error associated with the numerical differentiation performed is reported.
Test
Test Results
Link to Test Results page
Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI)
Computes the equilibrium crystal structure and energy for an arbitrary crystal at zero temperature and applied stress by performing symmetry-constrained relaxation. The crystal structure is specified using the AFLOW prototype designation. Multiple sets of free parameters corresponding to the crystal prototype may be specified as initial guesses for structure optimization. No guarantee is made regarding the stability of computed equilibria, nor that any are the ground state.
Test
Test Results
Link to Test Results page
Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI)
Computes the equilibrium crystal structure and energy for an arbitrary crystal at zero temperature and applied stress by performing symmetry-constrained relaxation. The crystal structure is specified using the AFLOW prototype designation. Multiple sets of free parameters corresponding to the crystal prototype may be specified as initial guesses for structure optimization. No guarantee is made regarding the stability of computed equilibria, nor that any are the ground state.
Test
Test Results
Link to Test Results page
Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI)
Computes the equilibrium crystal structure and energy for an arbitrary crystal at zero temperature and applied stress by performing symmetry-constrained relaxation. The crystal structure is specified using the AFLOW prototype designation. Multiple sets of free parameters corresponding to the crystal prototype may be specified as initial guesses for structure optimization. No guarantee is made regarding the stability of computed equilibria, nor that any are the ground state.
Test
Test Results
Link to Test Results page
Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI)
Given atomic species and structure type (graphene-like, 2H, or 1T) of a 2D hexagonal monolayer crystal, as well as an initial guess at the lattice spacing, this Test Driver calculates the equilibrium lattice spacing and cohesive energy using Polak-Ribiere conjugate gradient minimization in LAMMPS
Test
Test Results
Link to Test Results page
Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI)
Equilibrium lattice constant and cohesive energy of a cubic lattice at zero temperature and pressure.
Test
Test Results
Link to Test Results page
Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI)